
COMMENTARY ARTICLE
Beyond partnerships: redefining cdmo-biotech collaboration in the age of complexity
Operational Chemistry: Making Biotech–CDMO Teams Actually Work
Introduction
As the landscape of therapeutic development rapidly evolves, so do the operating models that support it. The relationship between biotechs and Contract Development and Manufacturing Organizations (CDMOs) has fundamentally shifted—from transactional outsourcing arrangements to strategic co-execution frameworks. In an era marked by increased scientific complexity, shorter Funding cycles, and increased regulatory scrutiny, there is no room for misalignment.
Advanced modalities such as antibody-drug conjugates (ADCs), oligonucleotides, and mRNA-based therapies require seamless integration across analytical development, quality systems, regulatory readiness, and manufacturing execution. At the same time, policies like the BioSecure Act highlight the need for operational transparency, supply chain traceability, and mitigation of geopolitical risks.
Despite these changes, many collaborations between biotechs and CDMOs still face issues like unclear expectations, fragmented decision-making, and reactive project management. Biotechs often enter partnerships expecting faster results and greater flexibility, while CDMOs struggle to interpret vague scopes within tight timelines—leading to operational conflicts, missed milestones, and loss of trust.
To better understand how collaboration models can be improved, this commentary relies on structured input from multiple biotech and CDMO leaders across the industry. Their responses to a curated set of operational and scientific questions highlight key patterns in both failure and success.
Specifically, this panel discussion explores:
- How biotechs assess CDMO fit for complex modalities
- How CDMOs engage with limited early-phase data to de-risk scale-up
- What practices foster trust, alignment, and decision clarity
- And how both sides can shift from coordination to genuine co-execution
The goal is not to repeat familiar challenges, but to suggest a practical operating framework for CDMO–biotech partnership—one rooted in mutual accountability, scientific consistency, and behavioral unity. In a sector where the pace of innovation now depends on operational discipline, collaboration has become a key skill.
From Vendors to Velocity Partners: Reframing CDMO–Biotech Collaboration
The conventional construct of the CDMO–biotech relationship — a transactional outsourcing model centered on deliverables and timelines — is no longer tenable in today’s high-stakes, modality-driven development environment. The increasing complexity of drug candidates such as antibody-drug conjugates (ADCs), oligonucleotides, and RNA-based therapeutics, combined with compressed development cycles and evolving regulatory expectations, demands a new framework: strategic co-execution.
In this model, CDMOs are not external executors but integrated contributors — scientific, operational, and regulatory partners accountable for shared success. The transition from “vendor” to “velocity partner” is not semantic. It is a fundamental redefinition of how biotech companies must approach outsourcing if they are to accelerate development without compromising quality or compliance.
Moving Beyond Siloed Execution
Legacy engagement models — often driven by procurement timelines or minimal technical criteria — fail to anticipate the ambiguity and risk that define early- and mid-phase development. Under such conditions, misalignment between sponsor expectations and CDMO operations leads to common and costly issues: rework, scope creep, tech transfer delays, and strained regulatory interactions.
Conversely, high-functioning partnerships exhibit the following characteristics:
Early-stage technical integration involving cross-functional input from chemical, finished dosage, analytical, quality, regulatory, and manufacturing teams.
Bidirectional accountability, where both parties share responsibility for meeting milestones and managing risk.
Operational transparency, backed by shared project governance and real-time data access
Unified interpretation of priorities, particularly around speed, quality, and cost under constraint
These are essential attributes for ensuring reliable execution amidst the rising demands of fast-to-clinic programs, not just aspirational ideals.
Values as Decision Infrastructure
Building on our previous work on organizational coherence, it is clear that values are more than just soft concepts; they serve as practical decision-making frameworks. When timelines are tight and information is limited, what guides CDMO and biotech team behavior? Is there a shared understanding of what to prioritize, when to escalate issues, and how to interpret “client-centric” or “risk-based” approaches?
Many breakdowns stem not from capacity or capability gaps, but from behavioral misalignment under stress. A CDMO may view “flexibility” as last-minute rework; a biotech might see it as proactive problem-solving. Without a shared language and decision logic, trust diminishes.
Effective CDMO–biotech collaborations, therefore, require aligned principles, not just aligned timelines. These principles must be embedded into operating rhythms — kickoff templates, governance models, risk registers — and reinforced in moments of ambiguity.
From Tactical Execution to Strategic Orchestration
CDMOs that succeed in this new paradigm act as orchestration partners. They proactively anticipate downstream regulatory implications, incorporate phase-appropriate controls, and guide biotechs through uncertainties. This role requires scientific fluency, regulatory literacy, and behavioral maturity to share ownership effectively.
Biotechs, in turn, must shift from outsourcing activity to co-designing execution — embracing clarity, readiness, and communication as core enablers, not overhead. CDMO–biotech collaboration must evolve from a supplier-based relationship to a strategic execution model grounded in alignment, transparency, and mutual accountability. This evolution is not optional. It is the price of speed, and the currency of trust in a biotech landscape defined by complexity and consequence.
What Biotechs Must Get Right: Clarity as a Competitive Advantage
While much focus is on CDMO performance and technical skills, a collaboration’s success or failure often starts earlier—in how biotechs prepare, set, and manage their own expectations. From the feedback collected from CDMO leaders across the panel, a clear pattern appeared: inefficiencies in early phases and problems in later execution are often caused by a lack of clarity from the biotech sponsor.
Precision Begins with the RFP
The request for proposal (RFP) sets the tone for the partnership, yet many CDMOs report that biotech RFPs are often aspirational rather than actionable. Common gaps include:
• Unclear project scope
• Incomplete analytical or process characterization data.
• Misaligned timelines relative to technical complexity
• Omission of phase-appropriate regulatory context
A robust RFP should clearly articulate not only the “what” (deliverables) but also the “why” (clinical objective, development phase, regulatory intent) and the “how” (critical quality attributes, control strategy, and risk assumptions). When this level of specificity is missing, CDMOs are forced to make assumptions that may later require costly realignment.
Biotechs that invest time in refining their RFPs—ideally with cross-functional input from analytical, regulatory, and CMC teams—set a more realistic foundation for the work ahead.
Internal Readiness and the Role of the Project Manager
Another common obstacle to efficient execution is the lack of a dedicated internal project manager (PM) with adequate scientific knowledge and decision-making power. Without this role, communication between the biotech and CDMO becomes delayed or scattered, and critical issues are either escalated too late or not addressed consistently.
Sponsors should ensure that the internal PM:
• Has cross-functional visibility (analytical, quality, clinical, regulatory)
• Maintains continuity of communication with the CDMO’s lead team
• Has the authority to approve scope clarifications and contingency decisions
The internal PM is not merely a coordinator but a key enabler of technical alignment and real-time decision-making.
Minimizing Scope Creep through Defined Milestones
Scope creep is a persistent challenge in CDMO–biotech engagements. In many cases, this results not from intentional expansion but from insufficient initial planning or evolving sponsor expectations.
To mitigate this, biotechs should:
• Define clear go/no-go decision points tied to data generation.
• Establish predefined buffers for non-critical change requests.
• Incorporate change control protocols into the governance plan.
Project charters should explicitly list what is in scope, what is excluded, and how changes will be assessed from both technical and budgetary standpoints.
CDMOs can only protect timelines if biotechs provide a stable scope of work supported by decision logic.
Building Tech Transfer Maturity
In early-phase collaborations, many biotechs struggle to assemble a complete and coherent technology transfer package. When methods are only partially qualified, or formulation data is preliminary, the burden of translation shifts to the CDMO—introducing risk, rework, and variability.
To improve transfer efficiency, biotech companies should:
• Use standardized tech transfer templates with predefined quality criteria.
• Provide method validation status and known method limitations.
• Include risk assessments with development history and troubleshooting insights.
Even when documentation is incomplete, transparency regarding the current state of knowledge allows CDMOs to plan development workstreams better and avoid duplication of effort.
Communication is a System, not a Meeting.
Finally, communication failure is rarely about frequency—it is about structure. Biotechs often rely on status meetings without a broader operating rhythm, leading to reactive behavior and decision paralysis.
Successful partnerships incorporate:
• Clear communication protocols with named points of contact
• Joint dashboards for real-time tracking of progress, deviations, and risks
• Phase-specific governance models that evolve with programming maturity
Sponsors must also ensure that internal escalation pathways are well-defined—so that urgent decisions do not get bottlenecked by indecision or lack of clarity about authority.
What CDMOs Must Do: Operating with Scientific Authority and Strategic Maturity
While much of the discussion about biotech–CDMO collaboration centers on sponsor readiness, CDMOs themselves must advance beyond just compliance and capacity rhetoric. In complex programs, especially those involving advanced modalities like ADCs, oligonucleotides, and high-potency APIs, scientific expertise alone is insufficient. CDMOs need to operate with strategic maturity—showing not only the ability to follow instructions but also the capacity to guide, anticipate, and co-create solutions in real time.
This section distills the expectations placed on CDMOs by biotech partners and outlines the institutional behaviors, systems, and capabilities required to meet them.
Engage Early with Scientific and Regulatory Perspectives
CDMOs that excel in complex programs consistently provide a technical voice early in discussions—during RFP review, feasibility assessment, and early protocol transfer. This scientific involvement is not just courtesy; it is a risk mitigation strategy.
Biotechs seek CDMO partners who can:
• Identify regulatory gaps or risks based on limited data packages.
• Translate early analytical signals into downstream manufacturing implications.
• Provide formulation or method development input aligned with the molecule’s lifecycle.
Failure to embed this perspective early results in downstream inefficiencies, scope renegotiation, or missed critical development windows.
Build Systems for Transparency and Traceability
In fast-moving collaborations, trust is built through data visibility and process traceability. CDMOs that rely on ad hoc updates, PDF-based communication, or siloed systems cannot meet modern expectations for responsiveness or control.
Leading CDMOs are now implementing:
• Real-time dashboards that provide shared access to batch progress, deviations, and documentation status
• Integrated LIMS and project portals that reduce redundancy and version conflicts
• Standardized tech transfer and method qualification templates accessible to both teams
• These systems not only improve delivery fidelity but also create a digital thread that supports audit readiness and regulatory defensibility.
Balance Flexibility with Phase-Appropriate Compliance
Biotechs often face tight deadlines—driven by funding milestones, clinical schedules, or board expectations. CDMOs need to respond to these demands without sacrificing compliance or scientific quality.
The hallmark of a mature CDMO is its ability to say:
“Yes, we can move fast — and here is how we’ll do it without sacrificing control.”
This requires:
Risk-based quality frameworks that adjust controls based on the development phase
Predefined “fast-track” pathways that align CMC activities with regulatory strategy
Scenario planning tools that identify bottlenecks before they occur
In this context, flexibility is not improvisation; it is disciplined agility, supported by structured decision logic.
Invest in Talent That Understands Modality Nuance
Complex modalities demand specialized scientific capabilities, including linker-payload chemistry, oligonucleotide synthesis, and conjugation platform development. More than equipment or SOPs, it is the depth and retention of scientific talent that defines a CDMO’s ability to manage risk, respond to ambiguity, and engage as a technical peer.
Best-in-class CDMOs demonstrate:
• Cross-trained scientists capable of integrating analytical, formulation, and process perspectives
• Structured onboarding and internal knowledge transfer mechanisms
• Visible career development pathways to retain niche talent in high-demand areas
• The biotech sector is watching not just what CDMOs build — but who they build with.
Communicate Risk Proactively and Constructively
A repeated point of frustration from biotech leaders is the lack of transparent communication around timeline risks. Delays are often inevitable in development. What separates competent CDMOs from strategic partners is how and when they communicate with them.
Proactive CDMOs:
• Share impact assessments before the sponsor asks
• Escalate based on triggers tied to timeline or quality thresholds.
• Propose mitigation strategies collaboratively rather than defensively.
• This approach not only protects trust but also improves decision speed and regulatory positioning.
• Lead Lifecycle Thinking from the Outset
Many biotech programs face fragmented planning—where early-phase decisions lead to complications on a commercial scale. CDMOs that adopt a lifecycle-integrated approach help sponsors make better trade-offs early.
This includes:
• Guiding phase-appropriate method validation with scale-up in mind
• Aligning formulation choices with long-term manufacturability and stability
• Advising on regulatory documentation that enables future bridging
CDMO’s role is not merely to deliver units; it is to de-risk development across the whole product lifecycle.
The expectations for CDMOs have changed. Sponsors now want partners who can smoothly integrate into their development ecosystem, provide scientific insights, ensure strict compliance, and advance progress with foresight. Operating at this level demands more than just technical skills; it requires strategic insight, transparency driven by data, and consistent reliability, even under pressure.
In a market driven by scientific ambition and operational constraints, CDMOs that serve as strategic co-developers—rather than just service providers—are the ones that consistently deliver value and build trust.
Shared Mistakes, Shared Lessons: Where Collaboration Breaks Down — and Why
Despite good intentions, CDMO–biotech partnerships often break down—not because of a lack of scientific knowledge or tools, but due to repeated behavioral and strategic mistakes. Input from CDMO executives and biotech leaders shows that common failure patterns exist. These lessons highlight that successful partnerships are rarely ruined by technical issues alone, but by unseen problems in alignment, ownership, and communication.
Misaligned Definitions of Success
One of the most common points of breakdown is the absence of a shared understanding of what success looks like at each stage of the program. While biotechs may prioritize clinical milestone acceleration, CDMOs may interpret “success” as compliance with specifications and audit-readiness. Without explicitly aligned definitions—embedded in governance charters and decision matrices—project teams tend to work at cross purposes.
Success criteria should be co-authored, not assumed. This includes technical endpoints, timeline tolerance thresholds, risk posture, and escalation paths. Alignment at kickoff is insufficient; it must be reinforced at each phase gate.
Scope Creep Without Structured Change Management
Scope creep is inevitable in development programs, particularly as additional assays, formulation modifications, or regulatory clarifications surface mid-project. However, many biotech sponsors expect flexibility from CDMOs without formally renegotiating deliverables, timelines, or budgets. This results in internal CDMO strain, timeline erosion, and eventual confrontation.
Change is not the problem; unstructured change is. CDMO–biotech agreements should include predefined change control frameworks with agreed timelines for re-evaluation, decision triggers, and impact assessments.
Delayed Communication of Risks and Deviations
A critical error—reported frequently by both CDMOs and sponsors—is the tendency to delay difficult conversations. Whether it’s a deviation that might affect a batch disposition, a slipped method transfer, or resource constraints due to concurrent programs, silence or soft communication of risk breeds mistrust.
The threshold for escalation should not be urgent, but potential impact. High-performing partnerships implement project risk registers with shared access and clear ownership for real-time status updates.
Poor Translation Between Functions
Scientific rigor often breaks down at the interface of functions—most notably between analytical and process development, or between regulatory strategy and operations. Many CDMOs and biotechs still operate in siloed functional hierarchies, causing rework, inconsistent documentation, and last-minute regulatory compromises.
Function-specific excellence is necessary, but insufficient. What matters is functional integration, which can be supported through cross-functional working teams, co-authored protocols, and shared accountability for deliverables.
Underestimating Cultural Compatibility
Finally, several panelists emphasized that cultural fit—the willingness to share information openly, the tolerance for ambiguity, the mutual respect for timelines and quality expectations—is a leading indicator of project success. Technical capability is essential, but without shared operating norms, even well-resourced projects can become combative and inefficient.
Sponsors and CDMOs must assess cultural alignment early, not post-contract. This includes expectations on responsiveness, documentation discipline, decision-making speed, and feedback culture. Pilot projects or limited-scope trials can serve as valuable diagnostic tools before expanding the engagement.
The most damaging failures in CDMO–biotech relationships are not dramatic—they are cumulative. They stem from assumptions left unspoken, risks left unflagged, and decisions left ambiguous. These failures are avoidable. When both parties institutionalize clarity, build systems for joint accountability, and create space for early tension detection, collaboration becomes not just functional but scalable.
The Missing Layer: Leadership as Operating Infrastructure
Technical competence and communication frameworks, while essential, are not enough to ensure robust CDMO–biotech collaboration. What often distinguishes successful partnerships is not how well systems are designed, but how leadership behaviors shape decision-making, risk management, and accountability under stress.
In high-complexity programs—particularly those involving novel modalities or accelerated timelines—uncertainty is inevitable. The ability to navigate that uncertainty depends on leadership, not as a function of title, but as an operating system. This includes how teams align, how decisions are made when data is incomplete, and how conflict is resolved without escalation.
These capabilities cannot be outsourced. Nor can they remain siloed. They must be embedded across the interface of sponsor and CDMO teams.
Leadership by Capability, Not Org Chart
Traditional collaboration models assign decision-making power by organizational hierarchy—biotech sponsors lead strategy, CDMOs execute tasks. But complex development demands a more fluid, capability-based approach.
Cross-functional mosaics, not individual experts, lead successful programs. These teams integrate regulatory foresight, analytical depth, quality mindset, operational fluency, and scientific agility into every project phase.
Key leadership capabilities include:
• Strategic translation: ability to connect clinical or regulatory goals with CMC execution
• Technical fluency across functions: particularly at the intersection of process, analytical, and formulation science
• Scenario planning and ambiguity tolerance: to adapt proactively under compressed timelines or shifting scope
• Ownership mindset: regardless of company boundary, functional role, or reporting line
From Meetings to Operating Models
Too often, “collaboration” is reduced to regular meetings and status updates. But meetings do not build alignment—systems do.
Transformative partnerships implement:
• Joint governance councils blending sponsor and CDMO leads across regulatory, analytical, and manufacturing functions.
• Shared accountability structures with co-owned milestones and KPIs
• Transparent escalation paths that empower frontline teams to resolve issues rapidly
• Integrated OKRs (objectives and key results) that drive shared behavior, not parallel effort
This is not about adding layers of oversight, but replacing reactive communication with proactive coordination mechanisms that scale.
Culture Is a Leadership System
Culture—how people behave when decisions are complex—is not the job of HR. It is a reflection of what leaders reward, tolerate, and model.
High-trust collaborations emerge when:
• Teams feel safe surfacing risks before they escalate.
• Leaders model transparency rather than defensiveness
• Constructive dissent is encouraged and integrated into decision-making.
• Feedback loops are fast, non-punitive, and anchored in shared outcomes.
These traits are not aspirational; they are now prerequisites for operating at the speed and complexity required by today’s biotech programs.
AI, Automation, and Digital Fluency as Leadership Leverage
As CDMOs and biotechs expand their digital infrastructure, leadership must evolve to own the application of tools such as LIMS, collaborative portals, predictive risk modeling, and proposal automation—not merely delegate them to IT or operations.
Digital maturity in a collaborative setting requires:
• Understanding how AI can augment forecasting, scenario planning, and stability modeling
• Leveraging dashboards and shared systems for visibility and accountability
• Ensuring that data quality and decision logic remain intact as speed increases
The future of development execution is digital—but only if leadership systems are ready to translate that capability into value.
True collaboration is not built on Gantt charts or signed SOWs. It is built on shared leadership behaviors that drive scientific integration, operational agility, and decision clarity. The absence of this layer—despite technical proficiency—often explains why promising partnerships fail.
Redesigning Collaboration: A Co-Execution Framework
In a development environment defined by speed, complexity, and constrained resources, the traditional CDMO–biotech model—based on service-level agreements and technical deliverables—has reached its limits. What’s needed now is a co-execution framework: an operating model that enables two distinct organizations to function with shared clarity, joint accountability, and adaptive execution.
This section outlines the foundational components of strategic co-execution, translating insights from both sponsor and CDMO perspectives into practical structures that enhance scientific integration, operational discipline, and long-term scalability.
Joint Operating Rhythm
A hallmark of effective collaboration is not how often teams meet, but how consistently they act across functions and phases. Co-execution requires a structured operating rhythm that aligns both companies’ activities without redundancy.
Core elements include:
• Phase-specific governance models (e.g., discovery, pre-IND, post-Phase I)
• Shared kickoff frameworks including roles, risk maps, and escalation logic
• Integrated calendar of activities tied to joint milestones—not just internal deadlines
This structure reduces project drift and prevents late-stage surprises.
Co-Owned Technical Risk Register
Most development programs will encounter scientific ambiguity, shifting priorities, or unexpected assay behavior. What differentiates high-performing teams is the ability to identify, document, and manage risk collaboratively.
A shared risk register should:
• Capture scientific, regulatory, and operational risks in a standard format.
• Assign joint ownership and mitigation actions.
• Serve as a live document reviewed in governance meetings.
• This approach normalizes complexity and moves teams from reaction to preparedness.
Bi-Directional KPIs and Incentive Alignment
Conventional CDMO performance metrics (e.g., on-time delivery, batch success rates) are necessary but insufficient. Sponsors also need insight into how work is executed—especially when timelines slip or scope evolves.
Co-execution introduces bi-directional metrics, such as:
• Sponsor responsiveness and scope stability
• CDMO deviation resolution speed and communication clarity
• Joint adherence to change control timelines
• Shared success indicators (e.g., IND filing quality, audit readiness)
• Including these metrics in QBRs and performance dashboards increases transparency and trust.
Digital Integration and Shared Systems
Co-execution is enabled by real-time, secure access to critical data. Many delays stem not from technical issues, but from fragmented systems and asynchronous communication.
Modern partnerships now employ:
• Shared dashboards (timeline, batch progression, document status)
• Collaborative document portals with version control
• Integrated LIMS access or tailored data exports for cross-validation
These tools reduce time lost to clarification, duplicate requests, and information silos.
Scenario Planning and Pre-Approved Contingencies
One of the most effective tools in complex program management is structured scenario planning. Teams that anticipate potential roadblocks—regulatory shifts, method delays, manufacturing constraints—are better equipped to respond without disruption.
Sponsors and CDMOs should jointly define:
• What-if scenarios across timeline, regulatory, and CMC variables
• Pre-approved contingency pathways (e.g., alternative assay validation strategies)
• Financial and timeline impacts for each scenario
• This minimizes reactive decision-making and maintains project momentum.
Behavioral Expectations and Decision Logic
The final layer of co-execution is behavioral. Teams must agree on how decisions are made under ambiguity, especially when speed, quality, and cost collide.
Best practices include:
• Documented decision-making frameworks for scientific ambiguity or deviations
• Clarified roles for technical, regulatory, and quality sign-offs
• Expectations around escalation etiquette, issue framing, and feedback cycles
These norms prevent internal friction and enable productive disagreement—a critical trait in innovation-intensive environments.
Strategic co-execution is not a theory. It is an evolving framework that builds resilience, clarity, and speed into CDMO–biotech partnerships. By aligning technical governance, risk transparency, communication infrastructure, and shared behavioral norms, organizations can collaborate more like integrated teams than independent contractors.
Closing: The Partnership Litmus Test
In an industry where timelines are non-negotiable, innovation is high-risk, and regulatory scrutiny is intensifying, the difference between a successful drug development program and a failed one often comes down to how well two organizations work together under pressure.
What has become evident through this panel—and through two decades of hands-on development leadership—is that traditional sponsor–CDMO relationships, based on compliance, contract terms, and tactical coordination, are no longer sufficient. The future belongs to partnerships grounded in strategic co-execution: joint decision-making, shared behavioral norms, and integrated risk ownership.
This shift is not just philosophical. It is operational, measurable, and increasingly essential.
The Litmus Test for Modern Collaboration
As both biotechs and CDMOs reflect on their current partnerships, a simple but revealing test can help evaluate maturity:
• When ambiguity arises, do both teams seek clarity—or seek cover?
• When timelines slip, do both sides adjust together—or escalate blame?
• When decisions are urgent, do both parties act with aligned judgment—or retreat into functional silos?
• And when success is achieved, is it viewed as mutual or one-sided?
If the answers to these questions expose hesitation, silos, or reactive behavior, then the partnership is not yet operating at its full potential.
The Real Differentiator Is Not Infrastructure — It’s Behavior
Many CDMOs today boast advanced instrumentation, expanded cleanroom space, and cross-site scalability. Likewise, many biotech firms bring cutting-edge science and promising therapeutic platforms. Yet execution still fails—not for lack of capacity or intellect, but for lack of behavioral alignment under stress.
What defines a high-performing collaboration is not the absence of problems, but the presence of:
• Clarity when priorities collide
• Transparency when risk emerges
• Shared logic when the data is incomplete
• And trust when speed and quality must coexist.
These are the traits that drive not only faster filings and more robust submissions—but also repeat business, team retention, and long-term credibility with regulators and investors alike.
Final Reflection
As complexity deepens and expectations rise, both CDMOs and biotechs face a shared imperative: to rethink how they partner — not just to deliver, but to learn, adapt, and succeed together.
Throughout this panel, leading biotech, CDMO executives shared candid insights from the frontlines — where ambitious science meets operational reality. What emerged wasn’t a single “right” model, but a recurring theme: alignment is everything.
The organizations that will define the next era of drug development won’t be those with the largest footprint or the fastest timelines. They’ll be the ones that master the art of thinking together, deciding together, and moving forward together — anchored in scientific discipline, operational clarity, and behavioral trust.
Because in today’s environment, execution is no longer the finish line.
It is the differentiator. Alignment is the new infrastructure.
–
Kishore Hotha, PhD, MBA
President, Dr. Hotha’s Life Sciences LLC
Dr. Kishore Hotha is a distinguished leader in the pharmaceutical biotech and CDMO sectors, with a strong track record in advancing drug substance and product development across small and large molecules, including Antibody-Drug Conjugates (ADCs), oligonucleotides, peptides, and complex formulations. Throughout his career, he has been pivotal in the submission of numerous INDs, NDAs, and ANDAs, guiding these projects from concept to commercialization. Currently, Dr. Hotha is the President of Dr. Hotha’s Life Sciences LLC, a consulting firm dedicated to simplifying complex drug development challenges. With over 100 publications and several editorial board positions, Dr. Hotha remains an influential figure in shaping industry standards and advancing pharmaceutical innovation.
–
Marie-Sophie Quittet
Head of Customer Relationships at Adragos Pharma, Jura
What should biotechs include in their initial RFPs to better align CDMO capabilities with project expectations?
A clear and detailed RFP is foundational to a successful collaboration. Sometimes, RFPs are vague or incomplete, leading to misaligned expectations or time-consuming clarifications later on. For an RFP to be effective in the area of fill-finish technologies, biotechs should include the following:
• Project scope and objectives: Clearly define both immediate and long-term goals (e.g., Phase 1 clinical supply versus commercial readiness).
• Timeline requirements: Outline all deadlines, including flexibility windows and any critical milestones for funding or clinical trials.
• Technical and regulatory requirements: List product characteristics (e.g., biologic modality, expected batch sizes, necessary technologies such as lyophilization or single-use systems, desired sterilisation method, security information such as cleaning and safety data sheets(SDS), regional regulatory constraints, and the preferred quality standards (e.g. FDA, EMA, JP).
• Material specifications: Share as much as possible about the API, excipients, packaging preferences, and whether ready-to-use components are preferred.
• Analytical methods and expectations: Be explicit about the analytical support needed, stability study design, and expectations for method development or transfer.
• Logistics and supply chain needs: Note any special requirements for storage, shipping, and material tracking.
Importantly, I would recommend sharing any uncertainties openly. For instance, whether certain analytical methods might not yet be fully validated, or if clinical timelines are dependent on funding. This transparency enables the CDMO to propose the best options and flag any feasibility or risk factors early on.
What steps can biotechs take to define project requirements better and avoid unnecessary delays?
Project delays in biotech-CDMO collaborations often stem from incomplete upfront alignment on what is required and when. Biotechs can save considerable time (and resources) by investing in thorough preparation before engaging with a CDMO. Key actions include:
• Internal alignment: Ensure all stakeholders (regulatory, clinical, supply chain) agree on the project’s objectives and constraints prior to engaging the CDMO. This reduces change orders and confusion later.
• Feasibility assessment: Where possible, conduct a pre-feasibility review (sometimes with input from your chosen CDMO) to identify technical gaps (e.g., need for filter validation, specific leachables studies) and regulatory issues.
• Prioritize requirements: Distinguish between essential and “nice-to-have” deliverables, particularly for early phase projects where resource efficiency is crucial.
• Define analytical requirements early: Many delays arise when analytical methods are unavailable or unsuitable for intended IPC (in-process controls) or release specifications; engage QC teams early and be explicit about what, when, and where analytics are needed.
• Prepare for material supply and readiness: Given current global supply chain risks, clarify with the CDMO lead times for critical materials (e.g., filters, excipients), and consider agreeing on safety stocks or backup suppliers.
At Adragos Jura, we have often accelerated timelines for clients who provide detailed material readiness plans, and proactively address potential gaps, such as defining appropriate volumes to discard post-filtration or aligning on buffer preparation methods in advance.
How can biotechs establish more apparent communication channels to avoid misalignment with CDMO teams?
Open, structured communication is the backbone of any successful biotech-CDMO partnership. Misalignment frequently occurs when communication is either too infrequent or routed through too many, or too few, channels. To mitigate this, I would recommend the following:
• Single point of contact: Nominate a dedicated project manager on the biotech side (ideally mirrored by a CDMO project lead). This streamlines decision-making and accountability.
• Kick-off meetings: Hold formal project kick-off sessions with all stakeholders present, including those from analytics, QA, production, logistics, and regulatory teams, to ensure a shared understanding of objectives, timelines, and critical process steps.
• Regular progress updates: Implement weekly or bi-weekly project calls, with agendas and action items circulated in advance. For complex or multi-site projects, shared digital project trackers (e.g., Gantt charts or dashboards) are invaluable.
• Clear documentation and traceability: Ensure that manufacturing and analytical records are available in real time to both parties, fostering transparency and enabling rapid response to unforeseen issues.
• Escalation pathways: Predefine how and when issues will be escalated, both for urgent operational matters and for strategic scope or regulatory questions.
Our most successful biotech clients at Adragos Jura are those who treat the partnership as a joint venture, not a one-way service, actively participating in regular reviews and openly flagging upcoming challenges or changes.
What advice would you give biotechs about balancing cost, quality, and timelines when selecting a CDMO?
Balancing the “iron triangle” of cost, quality, and speed is one of the critical strategic tasks for any biotech team engaging a CDMO, and it inevitably requires tailored trade-offs at different stages of development:
• Early phases (clinical supply): Prioritise flexibility and scalability over the lowest cost per vial. A CDMO with adaptable equipment and experienced aseptic staff may save months in tech transfer and troubleshooting later, even if unit costs appear higher.
• Later phases (commercial readiness): Quality and regulatory robustness come into sharper focus, as does the reliability of supply. Here, proven audits, robust QA teams, and global regulatory expertise (e.g., FDA, EMA, PMDA experience) become non-negotiable.
• Cost transparency: Insist on clear, detailed proposals from the CDMO that identify all deliverables, pricing models, and the nature of out-of-scope work. Avoid bundled pricing where possible; options à la carte encourage transparency and trust.
• Mutual risk management: Understand what risks (timeline, technical, supply chain) each party is bearing and clarify upfront how these are managed and mitigated. For critical activities, such as custom materials or unique sterilisation steps, ensure that “what-ifs” are discussed and costed.
• Invest in partnership: Both sides should be prepared for flexibility. At Adragos Jura, we accommodate accelerated schedules or documentation customisations for clients if needed, who are willing to share clear project priorities and mutual risks; this approach often results in both cost and time savings by focusing resources strategically.
In summary, a high-trust, collaborative relationship anchored on transparent communication and mutual understanding of risks and deliverables is far more likely to yield both successful clinical outcomes and sustainable partnership value.
–
Alastair Hay1, Jonathan Loughrey2
1. VP Peptides, Almac Sciences
2. Director of Chemical Development for Early Phase, Almac Sciences
How do you approach early-stage discussions with biotechs during RFP or feasibility phases?
Initial engagement depends on the detail provided ranging from brief emails to full technical packages. Clarifications are common, and a call is usually the best way to align scope. For complex projects, face-to-face meetings are ideal to ensure mutual understanding and accurate proposals.
What scientific support can CDMOs or CROs provide to biotechs during lead optimization and candidate selection?
Support is either transactional (e.g. FTE-based synthesis and analysis) or enhanced with specialised services which Almac Sciences offer, e.g. polymorph screening, solubility/stability profiling, and counterion screening for peptides. These help biotechs assess and refine candidate molecule effectively.
At what stage in drug discovery do you recommend CDMO involvement to ensure smoother downstream development?
The obvious answer is to say “as soon as possible”, but that doesn’t always suit the biotech’s model, and the CDMO must always show a degree of flexibility. It is important that the biotech knows their molecule and that may be best served by having internal resource with hands on experience of synthesising and using it. If all the biotech’s experience comes from outsourced data, then it can make it more difficult to resolve technical challenges.
How do you align your technical team’s input when biotechs bring discovery-stage molecules with limited characterization or formulation data?
Limited data is common. Fortunately, at Almac Sciences, we have tremendous breadth and depth of capabilities, and these can be tailored to the details of the technical package at quotation stage with recommendations for project scope. Building a characterisation package (e.g., pKa, pI, log P/D) is recommended as it helps guide formulation strategies aligned with the intended administration route.
What capabilities have you built to support the transition from hit-to-lead or preclinical discovery to IND-enabling studies?
Rapid synthesis and design input are key and since this phase of work is often investigative in nature, it is typically best supported by FTE programmes. Early formulation scientists can also use characterisation data to engineer properties like solubility and stability, adjusting buffers, excipients, and surfactants to meet dosing requirements.
What best practices help ensure scientific alignment between CDMO/CRO and biotech analytical, formulation, and process development teams?
Strong project management is essential. Almac Sciences fosters technical-technical communication between its team and sponsors. Project managers and technical leads collaborate closely to maintain alignment and ensure smooth execution.
How do you plan internal resources and capacity when handling multiple biotech clients in parallel without compromising timelines or quality?
A project manager is assigned after contract signature. Departmental managers allocate resources based on portfolio needs, with regular reviews to identify availability and pinch points. Almac Sciences’ strong interdepartmental relationships support efficient delivery.
How do you communicate timeline risks and mitigation strategies effectively to biotech partners?
Almac Sciences emphasises scheduling from project kick-off, identifying critical path activities and milestones. Risks are communicated early, including sponsor-side bottlenecks like document reviews. Transparency via regular update meetings ensures shared responsibility and proactive mitigation.
What practices do you follow to ensure transparency, accountability, and traceability in complex development programs?
Communication is key, and Almac Sciences’ culture is very much one of information sharing from both a technical and project delivery perspective. Regular project meetings, clear notes with agreed decisions and tracked actions are crucial to ensuring successful project delivery.
How do you support complex modality onboarding (e.g., oligonucleotides, ADCs, PROTACs) from early-phase biotech companies?
Onboarding what may be viewed as a complex modality is no different from what may be perceived to be a simpler one, ie a small molecule. An expert provider is exactly that – they have the technical and regulatory know-how and the internal capabilities – people, equipment, analytical instrumentation – to be able to deliver the project. New modalities often have specific considerations to consider that may not match small molecule experience, and care has to be taken that a fit for purpose approach is adopted.
What tools or digital platforms (e.g., dashboards, LIMS, collaborative portals) have improved real-time visibility and engagement with biotech clients?
Digitisation enhances collaboration. Client-specific portals streamline file sharing and document review. Electronic approval tools replace traditional methods, improving efficiency and visibility throughout the project lifecycle.
What’s your approach to method development and tech transfer when receiving incomplete or early-phase packages from biotechs?
Thorough technical assessment of the RFP ensures accurate proposals. Incomplete data will likely increase the required development time, but it’s not unusual that information will be partial, or there is guidance from the client that improvement is required. The key is communication and strong technical assessment so that we minimise the chance of surprises – no one likes those!
Some modalities lend themselves to general rather than product or process specific knowledge. For example, in peptide projects, it is uncommon for high levels of detail to be provided on a manufacturing process at the preclinical stage. Instead, general expertise and practises are applied and amended on a product-by-product basis.
How do you support “fast-to-clinic” timelines while maintaining regulatory compliance and phase-appropriate quality?
This is a very interesting area and one Almac Sciences has paid a lot of attention to. Personalised medicine, e.g. cancer vaccines, demands speed and tailored regulatory expertise as each product is “made to order”. GMP principles remain, but streamlined processes are applied following appropriate justification. For conventional drugs, phase-appropriate development and integrated services accelerate timelines.
How do you attract, train, and retain specialized scientific talent in high-complexity areas such as linker-payload chemistry, RNA synthesis, and high-potency API handling?
Operating in a niche modality would imply a smaller talent pool, but often these specialised areas can be sold on the basis of their uniqueness and exciting prospects. Recruitment should focus on core skills over narrow experience; internal training is expected, and flexibility in hiring ensures access to capable scientists who can grow into specialised roles.
What examples or lessons learned can you share from both successful and challenging biotech partnerships?
Empathy and collaboration drive success. Progressing a molecule from the bench to the clinic is an extremely difficult thing to do, and challenges will arise first time round. Strong relationships help navigate first-time challenges. Transactional partnerships struggle with problem resolution and understanding each other’s pressures fosters resilience and solution-oriented teamwork.
How do you assess and communicate your ability to support lifecycle development—from early-phase through to commercialization?
Almac Sciences has experience of taking products from pre-clinical right the way through to commercial, and others that transfer in at a later stage. The same fundamentals apply across the spectrum of projects – regulatory adherence is critical. The real expertise lies in navigating critical considerations, such as determining the starting point for Process Validation and shaping the GMP-compliant manufacturing strategy accordingly. Practical aspects, like ensuring compatibility with the required production scale, also play a significant role in successful progression.
What qualities or behaviours define a successful biotech partner from your perspective?
In my view, the most effective relationships form where the sponsor is engaged but not overbearing. Too close, and the CDMO will be smothered, too distant, and there is insufficient grasp of the true project status. The ideal sponsor possesses enough technical and regulatory knowledge to challenge and understand CDMO work. Mutual empathy and respect ensure productive collaboration and effective problem-solving.
–
Matthias Henz, PhD
Partner at Alpha Lyncis AG
Evaluation of CDMOs
During initial due diligence, how do you evaluate a CDMO’s quality management system?
The article is a concise, forward-looking blueprint for QMS appraisal forcing due diligence teams to inspect how quality is embedded, not just whether documents exist.
The author proposes a three-pillar framework to guide the evaluation of a quality-management system (QMS) at a CDMO:
First, regulatory compliance is verified through classic audit techniques, ensuring that all essential procedures exist and satisfy the minimum GMP requirements.
Second, the confirmation of a modern QMS design is done by looking for a modular procedure architecture, verifying the existence of lifecycle-aligned control strategies that grow from R&D to commercial supply and of built-in risk-based standardized approaches. Furthermore, procedures are supposed to be lean containing a series of strong visualization elements so that an independent reader is capable to understand the content of a procedure within five minutes- the “IKEA principle”.
Third, the depth of the embedded GMP culture and leadership governance is assessed by examining clear escalation pathways for significant events, data-driven KPI dashboards with documented follow-up actions, and management-review processes (Quality Council) that lead to tangible improvements.
The author believes that a CDMO demonstrating strength across all three pillars provides sponsors an enhanced assurance of sustained product quality and patient safety.
Traditional Due-Diligence Approach
In most CDMO due-diligence reviews, sponsors concentrate on the documented components of the Quality Management System (QMS) and on evidence of compliance adherence to procedures. Although essential, these document-centric review and compliance assessment provide a static snapshot and rarely illuminate whether the system functions dynamically enough to foster a mature quality culture. A robust due diligence program should therefore also scrutinize the QMS architecture and ascertain the depth of senior management’s day-to-day engagement in quality activities.
Modular QMS Architecture
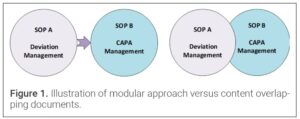
During a due-diligence review, sponsors should verify that the CDMO’s Quality Management System (QMS) is organized in modular architecture – discrete yet inter-linked policies and procedures that govern each functional domain. This structure delivers operational flexibility and procedural clarity, allowing individual SOP to be introduced, revised, or retired without disrupting the entire system. Consequently, improvements can be prioritized according to organizational maturity or emerging regulatory requirements, keeping the QMS agile and aligned with sponsor expectations.
Figure 1 illustrates why a modular design must also enforce clear boundaries between documents. The left panel depicts the desired state: two procedures with distinct, non-overlapping content. The right panel shows redundant content appearing in both procedures, which increases complexity and invites inconsistencies during future revisions – a risk that should be eliminated in any CDMO QMS. By defining precise remits and directing users to other procedures, rather than replicating related content, a modular framework safeguards against such content duplication.
Lifecycle-Driven, Risk-Embedded QMS Design
Modern QMS architecture should incorporate a lifecycle-based procedural framework that reflects GMP, compliance, and regulatory expectations in line with product maturity – from API starting materials (API-SM) and GMP intermediates to full-scale commercial APIs. Aligning procedures with each clinical and commercial phase guarantees compliance with ICH Q7 (including Chapter 19) and other prevailing standards (refer to Figure 2).

Within this framework, a CDMO must also anticipate the inherent, foreseeable risks that accompany its systems and processes. Whenever such risks affect quality, compliance, or operational integrity, control measures should be built directly into the relevant procedures. Embedding these “risk-designed” controls at the outset ensures that predictable hazards are managed systematically, prevents routine issues from escalating into deviations, and delivers consistent risk mitigation throughout the manufacturing lifecycle
Visual-First QMS Design: The Five-Minute Rule
A contemporary CDMO QMS should adopt a visual-first philosophy that accelerates comprehension, simplifies navigation, and enhances transparency. Under this model, any user – regardless of function or seniority – should be able to grasp the purpose, scope, and structure of a procedure within five minutes (“the Five-Minute Rule”).
To achieve this, policies and procedures are augmented with purpose-built visual aids:

This approach mirrors the picture-led instructions “IKEA Principle”: complex furniture assemblies are distilled into clear visuals that let untrained individuals build reliably the first time right. Translating that principle to pharmaceutical operations replaces text-heavy procedures with visualization aids, markedly avoiding misinterpretation and deviation while sustaining full GMP compliance and reducing cost.
Management-Engagement for Proactive Quality Governance
During due diligence, the sponsor should not only verify the existence of governance procedures but also how effectively they are embedded in daily practice. A robust CDMO QMS therefore includes dedicated Management-Engagement related procedures that make senior leadership directly accountable for quality performance, risk-based decision-making, and continuous improvement. Key elements include:

These procedures and their application empower management to track performance in real time, address emerging issues before they escalate, and ensure that quality objectives remain tightly coupled to corporate goals – hallmarks of a mature QMS at a CDMO, which fosters confidence at the sponsor.
–
Diego Schmidhalter
Partner and Director at Alpha Lyncis AG
Technology Transfer – More than Handing over Paper
How do you assess and communicate your ability to support lifecycle development from early-phase through to commercialization?
All partners of Alpha Lyncis have more than 25 years of experience, mostly in the CDMO industry, in various senior operations and business management functions in R&D, MSAT, manufacturing operations, quality, technical innovation and business development. This enables our experts to provide broad strategic and technical support over a majority of relevant business areas and business processes and over a broad range of production technologies such as for microbial, cell-culture based products, chemical synthesis of APIs, peptides and oligonucleotides, ADC, mRNA, biotransformation, biocatalysis and others.
Our customer base includes start-ups, SMEs and large pharma companies, including CDMOs. The focus of our service is on enabling our customers to be more effective and efficient, by identification and implementation of best-in-class process solutions, accelerating process development and de-risking programs and technology transfers through early identification of pitfalls and risks. Other focal points include the development and implementation of a holistic strategy for single-use technology, definition of equipment lay-out, the modular implementation of a turnkey quality management system and the use of our extensive professional network to bring together project partners.
At what stage in drug discovery do you recommend CDMO involvement to ensure smoother process development and what services can a CDMO provide?
Focus of drug discovery is on identifying a product, showing superior performance in a treatment or in the prevention of a disease. CDMOs typically enter the stage when it comes to develop a production process, suitable for future commercial manufacture which, in the case of cell-based production, comprises strain or cell-line development.
The choice of a best production organism is paramount. This is where experience of a CDMO comes into play. Leading CDMOs offer access to superior expression technology in combination with high-throughput (HTP) screening and HTP process development capabilities. This combination allows to significantly speed up the development of clones, selection of a best producing clone, fermentation or cell culture and purification development. Unlike most biotech companies, CDMOs work on a large number of products within a one-year period, which significantly adds to the expertise of CDMO scientists allowing them to develop high-titer USP processes and high-yield DSP processes while delivering a product that meets specifications.
mRNA products are typically produced using an in-house developed platform process. Therefore, mRNA companies are approaching CDMOs mainly for the production of pDNA or for the manufacture of product i.e., production of mRNA bulk substance and final formulated bulk drug.
Due to the high risk involved, biotech companies are advised to choose a CDMO partner very carefully as cost for developing a product are considerable and high regulatory standards must be met. Entering into a collaboration is usually the start of a long-term relationship, demanding a good cultural fit. A defined list of key selection criteria, previous experience and knowledge of the CDMO sector at a global level, including an understanding of the associated costs, are important prerequisites for selecting the best possible partner.
What best practices help ensure scientific alignment between CDMO/CRO and biotech analytical, formulation, and process development teams?
There are numerous practices that have a positive impact on the alignment between the project sponsor’s team and the service provider’s team. To mention just a few:
- Open communication and mutual trust are key. It begins as early as the RFP-Proposals phase. Already in this phase the two teams should be connected to ask and answer question to develop a mutual understanding of the current status of process and analytical development and the properties of the product. A thorough assessment helps to determine the starting point and to verify the deliverables.
2. A detailed project plan based on standardized work-flows should list project deliverables and timelines.
3. A clear project team structure, led by program managers on both sides, supports smooth interaction between the teams as well as having an escalation process in place. This protects scientists from being drawn into stressful discussions they have no decision-making authority.
4. Weekly cross-team meetings, to share results and discuss observations.
5. Process development scientists and an analytics expert of the sponsor should be present in the CDMO’s laboratories during the technology transfer, i.e. to support the first verification batch(es).
6. Technology transfer should base on a structured approach. Overall, we made good experience with following a two-level stage gate process, a first thorough assessment of the project development status after finalization of the initial verification batches to address potential changes to the program and a second stage gate after completion of the agreed program to assess production readiness. Examples of aspects to asses during the stage gate reviews are: robustness and reproducibility of each unit operation, product stability, availability of analytical methods, raw materials and biosafety, EMC, safety challenges, equipment suitability and cleanability and process economy.
7. I also advice the use of visual standard work plans to reduce friction in a complex working environment, such as tasks requiring collaboration of multiple departments, campaign preparation being an example.
What tools, digital platforms have improved real-time visibility and engagement with biotech clients?
Visual as well as digital tools support communication on different levels between the sponsor company and the CDMO. Scientists and program managers focus on adherence to the detailed project plan and progress against that plan. Upper management wants to understand the overall status of a project, with issues and risks highlighted. This can be perfectly visualized in a one-page project dash-board. Some CDMOs are sharing project-specific real-time data by giving access to their global data mart or LIMS.
What lesson learned can you share from both successful and challenging biotech partnerships?
The relationship between sponsor and CDMO is a collaborative effort based on trust. Therefore, maintaining a good relationship between the involved is of crucial importance. This includes maintaining a positive attitude, assuming that both parties are striving for the best possible project success. Trust cannot be taken for granted. So, it is important to immediately communicate issues and incidences, however unpleasant the message may be, whether the issue is due to force majeure or improper action – a rule, that is often not followed.
–
Dr. Chandrakanth Gadipelly
Principal Research Scientist & Co-Founder – Amar Flow Laboratory
Amar Equipment Pvt. Ltd. & Amar Flow Laboratory LLP
Biotech-CDMO Collaboration: A 2025 Perspective of a FlowChemistry Veteran
By 2025, the biopharmaceutical industry will showcase unprecedented diversity, including antibody-drug conjugates (ADCs), therapeutic oligonucleotides, mRNA vaccines, gene-editing payloads, and next-generation peptide conjugates. This expansion is invigorating innovation but simultaneously intensifying competition for specialized Contract Development and Manufacturing Organization (CDMO) capacities. Challenges compounded by fragile global supply chains, stringent regulatory scrutiny, and investor-driven urgency for rapid “idea-to-IND” timelines have transformed traditional outsourcing into deeply integrated strategic partnerships.
Key Structural Shifts Influencing Collaboration
Three pivotal structural shifts have emerged:
- Convergence of High-Hazard Modalities: New therapeutic modalities often involve potent toxins, hazardous chemical linkers, and complex synthetic intermediates. For example, ADCs incorporate high-potency payloads; siRNAs use fluorinated phosphoramidites; CRISPR therapies require precise synthetic controls. Consequently, advanced containment systems and highly automated micro- and meso-flow reactor technologies are imperative to safely handle these substances.
- Accelerated Regulatory and Digital Expectations: The formalization of continuous manufacturing through regulatory frameworks like ICH Q13 and initiatives such as the FDA›s Framework for Advanced Manufacturing (FAM) have significantly increased demands for stringent data integrity and real-time batch release capabilities. Sponsors now anticipate CDMOs to provide transparent, compliant electronic batch records with real-time monitoring.
- Geopolitical and ESG Pressures: Government-led reshoring incentives in the US and EU, alongside India’s Production-Linked Incentive (PLI) schemes, are reshaping global capacity distribution. Moreover, Environmental, Social, and Governance (ESG) criteria, particularly Scope-3 carbon emissions reporting, prioritize solvent minimization, single-use flow equipment, and renewable energy sources.
Navigating this landscape demands meticulous partner evaluation, realistic scheduling, disciplined governance, and transparent communication between biotechs and CDMOs from the outset.
Critical CDMO Evaluation Criteria in 2025
To select suitable CDMO partners, biotechs should prioritize:
• Modality-Specific Technological Depth: Ideal CDMOs demonstrate robust continuous-flow capabilities for high-potency active pharmaceutical ingredients (HPAPI) validated at over 10 kg/year scales, equipped with inline analytical monitoring systems like FTIR and UHPLC. Specialized purification techniques such as tangential-flow filtration, dual-column chromatography for ADC payloads, and enzymatic capping modules for oligonucleotide synthesis are essential benchmarks of advanced technological depth.
• Regulatory and Quality Maturity: Top-tier CDMOs exhibit impeccable regulatory records, demonstrated by zero repeat observations in recent FDA or EMA inspections, robust electronic Quality Management Systems (e-QMS) compliant with ALCOA-plus principles, and active participation in regulatory innovation programs such as FDA›s FAM pilot.
• Operational Flexibility and Buffer Capacity: Facilities should feature modular cleanroom setups capable of rapidly transitioning between product modalities (e.g., cytotoxic compounds to oligonucleotides) within short turnaround times, supported by well-trained, multidisciplinary teams. Stable staffing with turnover rates below 10% ensures consistent quality and continuity.
Validating CDMO Performance and Fit
Biotechs can validate CDMO capabilities through:
• Reviewing multiple randomly-selected project schedules (Gantt charts) for timeline adherence, noting cumulative delays as indicators of reliability.
• Confirming Right-First-Time (RFT) batch success rates above 90%, where lower scores indicate potential quality or regulatory issues.
• Inspecting real-time Process Analytical Technology (PAT) implementations and Statistical Process Control (SPC) charts, as live data streams demonstrate genuine process control versus static, potentially misleading snapshots.
• Conducting thorough quality audits by tracking deviations through Corrective and Preventive Action (CAPA) processes, verifying floor-level implementation within defined timelines (ideally under 30 days).
• Assessing regulatory compliance histories, interpreting isolated versus recurring compliance issues, and reviewing active regulatory engagement such as ongoing agency interactions.
• Evaluating infrastructure alignment, ensuring redundancy in utilities (e.g., chillers, air systems), appropriate containment for hazardous materials, and correctly scaled, validated equipment for intended production volumes.
Additionally, CDMOs provide critical scientific support during the lead optimization phase by aiding in identifying scalable synthesis routes, robust analytical method development, and stability evaluations, thus significantly mitigating downstream scale-up risks.
Ensuring Timely Execution
Robust scheduling requires detailed bottom-up planning based on historical data, clear identification of critical paths, proactive incorporation of realistic raw-material lead times (e.g., 20 weeks for phosphoramidite procurement), and contingency planning, including dual-sourcing strategies for critical inputs. Transparent and proactive communication regarding potential risks and implementation of Joint Steering Committees (JSCs) facilitate quick resolution and alignment.
Key performance indicators (KPIs) such as schedule adherence (>90%), deviation closure timelines (<30 days), and Batch Record accuracy (>95%) offer predictive insights into potential project risks. A disciplined project management approach, including clearly defined hold-points and liquidated damage clauses for delays, ensures balanced speed and compliance.
Practical Recommendations for Biotechs
• Construct detailed Requests for Proposals (RFPs) clearly outlining Quality Target Product Profiles (QTPP), anticipated production scales, known hazards, and intellectual property positions.
• Pre-contractually define Critical Process Parameters (CPPs) to control scope effectively and manage change requests via formal documentation systems.
• Establish centralized digital communication platforms for real-time project updates, including visual status indicators and regular briefing sessions.
• Conduct rigorous technology transfer preparations, providing comprehensive documentation and performing gap assessments well in advance of transfer initiation.
• Engage dedicated internal project managers versed in chemistry, regulatory compliance, and finance to effectively oversee CDMO collaborations.
• Pilot small-scale feasibility studies to assess technical capability alignment, communication effectiveness, and cultural compatibility prior to full-scale engagements.
• Use balanced decision-making matrices, weighted towards quality (40%), timeline reliability (35%), and cost considerations (25%), rather than defaulting solely to lowest-cost options.
Conclusion
While complex therapeutic modalities represent substantial clinical opportunities, they simultaneously elevate manufacturing and regulatory risks. Effective collaboration between biotechs and CDMOs is thus crucial. Front-end diligence—thoroughly vetting partner capabilities, ensuring real-time analytical proficiency, assessing regulatory maturity, and fostering transparent communication—represents the single most critical investment in securing timely, successful drug development. Meticulous evaluation and careful planning mitigate risks, ensuring robust operational performance and avoiding costly delays. In 2025’s fiercely competitive biopharmaceutical landscape, such comprehensive due diligence and transparent collaboration are not just advisable; they are imperative for sustained commercial and clinical success.
–
Subas Sakya
Chief Scientific Officer, BioDuro
How do you approach early-stage discussions with biotechs during RFP or feasibility phases?
At BioDuro, early-stage engagement typically begins with assigning a project manager upon receipt of an RFP. This person coordinates internal efforts to collect detailed responses from the relevant scientific and operational teams. The compiled information is reviewed, validated, and shared with the biotech partner. These discussions often continue as needed to clarify scope, address technical considerations, and ensure alignment on feasibility and expectations.
What scientific support can CDMOs or CROs provide to biotechs during lead optimization and candidate selection?
BioDuro supports biotechs during lead optimization by contributing to compound design, synthetic route development, biological evaluation, and DMPK profiling. In addition, our platforms with catalyst screening, photochemistry, flow chemistry and library synthesis enablement allows for rapid problem solving to move projects forward.
Single site location for cross-functional collaboration between discovery chemistry, biology, and pharmacokinetics allows for reduced cycle time and timely data generation, allowing for rapid decision-making. This integrated approach, with a single project manager POC, also enables efficient progression from lead optimization to preclinical candidate selection, including the ability to conduct in vivo PK and PK/PD studies when appropriate.
At what stage in drug discovery do you recommend CDMO involvement to ensure smoother downstream development?
Involving a CDMO like BioDuro at the stage of preclinical candidate selection is often beneficial. This is the appropriate time to introduce the compound to the process development and pre-formulation teams, who can begin evaluating manufacturability and formulation strategies. Early involvement helps reduce later-stage risks and supports a smoother handoff into IND-enabling studies.
How do you align your technical team’s input when biotechs bring discovery-stage molecules with limited characterization or formulation data?
When a discovery-stage molecule is introduced with minimal data, BioDuro’s technical teams assess whether additional characterization is needed. Common activities include solubility profiling, salt and polymorph screening, and evaluation of other physical properties. These steps help determine appropriate formulation strategies. Early pre-formulation and formulation screening also enable execution of in vitro and in vivo studies, which are critical at this phase of development.
What capabilities have you built to support the transition from hit-to-lead or preclinical discovery to IND-enabling studies?
BioDuro has developed integrated capabilities across discovery chemistry, biology, and DMPK to facilitate a seamless progression from hit validation through to candidate selection. At the point of preclinical candidate nomination, our process and formulation development teams are brought in to begin planning for scale-up and downstream development. This structured handoff allows for continuity across project stages. In one representative program, over 400 compounds were synthesized and tested within 16 months using this integrated workflow.
How do you plan internal resources and capacity when handling multiple biotech clients in parallel without compromising timelines or quality?
BioDuro manages resource planning by assigning dedicated project teams to each biotech partner. These teams include scientific staff, group leaders, and directors who are aligned with project needs and timelines. We have multiple sites for chemistry where clients can locate their teams along with needed analytical teams.
If integrated support across chemistry, biology and DMPK are needed, we have two sites enabled for cross functional collaboration. Each of these sites can support multiple clients with dedicated labs for IP security.
Having a large and experienced scientific workforce allows for flexibility and scalability while maintaining delivery standards across concurrent programs.
How do you communicate timeline risks and mitigation strategies effectively to biotech partners?
Timeline projections at BioDuro are based on scientific assessment and past experience. If risks are identified—such as potential delays from complex chemistry or external shipping issues—clients are informed early, and backup strategies are discussed. Open and ongoing communication is central to this process. The goal is to work collaboratively with biotech/pharma partners to manage uncertainties and maintain alignment throughout the project lifecycle.
What practices do you follow to ensure transparency, accountability, and traceability in complex development programs?
BioDuro maintains transparency through regular updates, including weekly meetings, detailed written reports, and ad hoc discussions when needed. Teams operate with a strong sense of ownership and responsibility. All scientific activities are recorded in lab notebooks and electronic systems in accordance with regulatory expectations. Internal QA audits and client inspections are routinely conducted to ensure traceability and compliance throughout the development process.
–
Sylvia Wojczewski, PhD
CEO, BioSpring GmbH
Partnering With The Right CDMO To Accelerate Clinical & Commercial Development Timelines
How do you approach early-stage discussions with biotechs during RFP or feasibility phases?
At all stages, beginning with the RFP process, communication is key when interacting with our biotech partners. Clear alignment on goals, objectives, and milestones define the success of a partnership. As a CDMO, we are responsible for understanding client and program-specific requirements to ensure we meet their desired timelines, deliver high-quality products and services, stay within budget, and support navigation of complex regulatory landscapes. Part of our role is to advise and guide on strategies tailored to the individual goals of the biopharma client. Early and rigorous planning allows us to work together to scale production and serves as a foundation for successful later stage development.
What scientific support can CDMOs or CROs provide to biotechs during lead optimization and candidate selection?
At what stage in drug discovery do you recommend CDMO involvement to ensure smoother downstream development?
Our goal is to engage with our clients as early as possible in the drug development lifecycle. Ideally, supporting them already during their lead candidate screening and optimization stage. During those early stages, it is key to be transparent as a CDMO. Sharing feedback concerning manufacturability of lead candidates as opposed to just delivering a product has helped many clients make informed decisions during their lead candidate selection process and address potential challenges early.
Further, as a CDMO with capabilities spanning from early development through commercialization, we leverage clinical and commercial manufacturing experience when working with our clients to prepare for the next steps, e.g., entering clinical development or additional scale-up and process refinement stages in preparation of commercial supply. Clients trust the proven concepts and expertise we apply consistently across all CMC activities. Considering scalability early on is essential to manage cost and timelines to secure market leadership early in this fast-paced biopharma environment.
As time-to-clinic, or time-to-market, is always crucial due to the increased competitiveness in the biopharmaceutical sector, collaborating closely with the CDMO partner early on helps to save valuable time and money when entering into clinical trials or commercializing a therapy. Working with a CDMO that is highly experienced with the given modality is critical, as this allows the CDMO partner to leverage its existing platform knowledge, ensuring ambitious timelines and tight budgets are met.
What capabilities have you built to support the transition from hit-to-lead or preclinical discovery to IND-enabling studies?
BioSpring takes a comprehensive approach to accelerating clients through the entire drug development lifecycle, with services spanning from manufacturing and regulatory support to analytical/bioanalytical services. We offer high-throughput production of nucleic acids to support discovery, then perform a seamless scale-up of the lead candidates to larger batch sizes for early preclinical work and regulatory toxicity studies. In parallel, we offer bioanalytical assays and a full suite of in-house analytical testing that provide more insight into the metabolic profile and characteristics of the therapeutic candidate. When the client is ready, we move with them through the clinical and commercial milestones, manufacturing cGMP batches and performing release and stability testing for both drug substance and drug product. This entire process is coupled by full regulatory support, such as scientific writing of dossier sections, and an expert panel of scientists from every department involved.
How do you support complex modality onboarding (e.g., oligonucleotides, ADCs, PROTACs) from early-phase biotech companies?
We are a CDMO specialized in manufacturing nucleic acids, with almost 30 years of experience. This experience also includes complex molecules involving customized chemistries and starting materials, and heavily modified and conjugated compounds. When onboarding new molecules and programs, we always aim to leverage existing platform knowledge to reduce onboarding timelines and hence time-to-clinic or time-to-market. We have dedicated teams for certain molecule types that help to ensure efficient knowledge transfer, ensuring flexibility and scalability to meet diverse manufacturing needs.
How do you communicate timeline risks and mitigation strategies effectively to biotech partners?
What practices do you follow to ensure transparency, accountability, and traceability in complex development programs?
Timeline risks and any technical challenges that might affect the success of a program need to be communicated imminently. Biopharma clients are under a lot of pressure and need to be aware of any potential risks immediately, considering that there are patients awaiting these life-changing treatments. Therefore, it is important to partner with a CDMO that can ensure full-transparency. We implemented best practices to guarantee full visibility and traceability at every stage of the project. Frequent project team meetings, as well as ad hoc meetings are important to keep all stakeholders informed and to ensure efficient resolution of any hurdles.
How do you support “fast-to-clinic” timelines while maintaining regulatory compliance and phase-appropriate quality?
As a CDMO, we know that heightened regulatory demands can complicate and slow the path to clinic.
Clients are often collaborating with us early on in preparation of regulatory filings for both their clinical and commercial programs. It’s important to clients that we demonstrate a high-level of flexibility and communication to accommodate their goals and tight timelines, as well as have robust workflows and capacities in place to ensure regulatory compliance and quality across all phases. Beyond providing manufacturing and analytical services, we also prepare our clients to be “regulatory-ready”. With our experience spanning almost three decades across a diverse client base, programs, and types of modalities, we are uniquely positioned to support clients with any regulatory questions and help them navigate this complex regulatory landscape while adhering to the highest quality standards.
–
Andrew Mitchell
Associate VP, Business Development, BIOVECTRA
At what stage in drug discovery do you recommend CDMO involvement to ensure smoother downstream development?
As one of the largest North American biologic microbial fermentation CDMOs with various available scales, we recommend biotech companies engage a CDMO as early as possible, especially if they anticipate potential issues during process development. Ideally, any issues will be identified at the preclinical or, at the latest, by the end of phase 1 clinical trials. With our experience of various projects, this timing allows us to make changes and finalize the process before phase 2, when having a robust and reproducible process is critical. When a biotech approaches CDMOs much later in the cycle, it’s likely going to add cost and involve some rework and timeline delays.
There might be a step that works well at a small scale in the lab —such as the chosen cell line or microbial expression system for a recombinant protein or plasmid— but won’t scale up efficiently for clinical trial production. Even when a startup is keen on getting phase 1 safety data as quickly as possible, knowing that most products won’t make it to the later phases, we proactively flag steps in the process that may cause problems during scale-up.
Another reason to engage early is that we may need to optimize our equipment based on the client’s process or their product. If, for instance, the process has an unusual step requiring a high volume of solvent or aqueous media, we need to confirm that we can handle those requirements or find an alternative solution. Early engagement allows us to match a biotech’s needs with our capacity. If they come to us during later stages of development, we may need to make adjustments to the process train equipment, which always has the potential to cause delays.
What’s your approach to method development and tech transfer when receiving incomplete or early-phase packages from biotechs?
We evaluate the initial package provided by the client for incomplete information. A follow-up meeting with the client and our technical team clarifies outstanding details. Our initial proposal includes assumptions—expected yield, recovery rates, and specific equipment requirements—and then we address any remaining unclear or missing information.
Once we win a proposal, we immediately do a familiarization run to ensure the package works as intended. This familiarization run may tell us we need to do further development work on a part of the process to scale up or, in rare cases, it may even indicate that we can’t go any further.
Each of our development hubs—for biologics, small molecules, and purification—supports tech transfers to manufacturing operations. The process development and manufacturing science and technology teams work in close coordination to minimize scale-up issues as processes are developed from early-phase packages for large-scale production.
What examples or lessons learned can you share from both successful and challenging biotech partnerships?
The success of a partnership depends on transparent communication. As a CDMO, we rely on clients to openly share what they know—as well as what they don’t. On our side, we have to explain any potential issues we foresee. Agreeing on expectations, deliverables, and the scope of work helps form the foundation of a productive collaboration and make a project successful. Once we’ve onboarded a project, the assigned project manager meets with the client every week for a project review and to ensure the collaboration is going smoothly.
Even then, there are ways communication can break down. Initially, we deal with the purchasing department of a biotech company. Once a project is onboarded, we tend to interact with project groups and transfer groups. If they work in isolation from their manufacturing groups, they may not understand the complete picture. When clients aren’t upfront about their challenges and provide vague expectations or targets without clear specifications, it can make it difficult for us to effectively deliver a product. A good example was a tech transfer of a continuous fermentation process with a pull-off on the tank that allowed us to complete downstream processing in parallel. We found the fermentation challenging to complete, prolonging the process. While we collaborated with our client to improve the aseptic processes and solve the problems, we learned they were facing the same issues internally—information that, if shared earlier, could have accelerated our problem-solving.
How do you assess and communicate your ability to support lifecycle development—from early-phase through to commercialization?
Our experience offering complete lifecycle support allows us to guide clients through each development phase. We help them understand how GMP requirements change for each phase of clinical trials as they become progressively more encompassing.
There are times when we assess a project and identify a gap between the client’s long-term needs and our existing capabilities. In these instances, we have a discussion about what’s needed to take their product to the volumes they require.
We have had several projects where we’ve successfully built capacity for a client’s specific program.
We are continuously reviewing and assessing our internal capabilities so we can best support clients. A good recent example is the expansion of our process development offerings. We now have a lot more equipment and the ability to support a larger number of projects side-by-side on the journey through the clinic to commercial. We now support clients in the lab from 250 mL to 50 L scales. Large-scale production for clinical material and beyond occurs in a commercial facility with larger equipment, including 100 L and 1,000 L single-use fermenters and 17,000 L stainless steel fermenters.
–
Kathryn L. Ackley, PhD.
Oligonucleotide CMC Consultant, USA
What are your top 3 criteria when selecting a CDMO for complex modalities like ADCs or oligonucleotides?
My top three criteria include the technical expertise of the CDMO staff, the CDMO’s analytical development capabilities, and the CDMO’s quality management system. The CDMO should have a team of people with experience in the modality of interest. If all the CDMO’s technical knowledge resides within a few people, the project will be vulnerable to staffing changes. Early in the vendor selection process, ask for a video call or on-site visit with the technical team that will oversee the project. The biotech may wish to seek assistance from a knowledgeable consultant to help with the vendor selection process. Consultants can be especially useful when the biotech’s internal staff has limited experience with the modality. Ask to see an organizational chart to verify that the CDMO has adequate staffing levels to support the project.
Complex modalities typically require complex analytical methods. Large molecules such as oligonucleotides often need orthogonal purity methods, multiple identification tests, and characterization of complex impurity mixtures. Significant analytical development resources are needed to support these activities. The success of the manufacturing process development depends upon analytical development. The manufacturing process is established by measuring the impact of changes to process parameters. The speed at which the CDMO can generate quality analytical data to support process development, manufacturing, and product characterization will dictate the pace of the entire project. For this reason, I always pay close attention to the CDMO’s analytical development capabilities.
Finally, the CDMO must have a robust quality system. A drug sponsor never wants to recall a drug due to safety concerns. Many drug sponsors wait to perform a quality audit after the vendor selection process is complete. The quality audit is often viewed as a formality, but the audit will uncover important information that can help with the vendor selection process.
What steps can biotechs take to define project requirements better and avoid unnecessary delays?
The first step a drug sponsor should take is to perform a thorough assessment of the company’s capabilities to identify areas where there may be gaps in expertise or resources. The drug sponsor is ultimately responsible for a drug’s safety. The sponsor may outsource activities such as manufacturing, but the sponsor must still provide sufficient oversight. The drug sponsor can obtain the necessary resources through hiring employees or through retaining consultants or contractors. The CDMO relationship is a partnership, and both parties need to have adequate resources to keep the project on track.
Make sure all the necessary internal stakeholders are involved in the initial project planning. The non-clinical, CMC, and clinical team members should all have the chance to review the project plan and make their respective needs known. Ideally, this review will take place before the RFP is finalized and sent to the CDMO. Project delays can occur when internal stakeholders have not been properly consulted and the project scope must be changed during the execution of the project.
Drug development programs generate many documents that require client review and approval. The CDMO will ask the drug sponsor to review documents such as development reports, manufacturing batch records, analytical protocols, and validation reports. These documents need to be reviewed by the client within a few working days to maintain the project timelines. The drug sponsor should develop an internal process for reviewing and approving documents from the CDMO before the project starts. Doing so will avoid delays.
During initial due diligence, how do you evaluate a CDMO’s quality management system?
A quality audit conducted by an experienced auditor is invaluable. Ideally the auditor will have experience with the type of drug and with the regulatory requirements for the region of interest. However, conducting an audit during the initial due diligence phase may not be feasible. In that case, the drug sponsor can still perform an initial evaluation. The CDMO should be able to provide the company’s regulatory inspection history and the outcomes of those inspections. Some countries grant licenses for GMP activities, and the CDMO should be able to provide this documentation upon request. Prior to GMP manufacturing, a quality agreement between the drug sponsor and the CDMO is put into place. Many CDMOs will provide a potential customer with the template for the CDMO’s standard quality agreement. The level of detail in the CDMO’s quality agreement will provide insight into the CDMO’s quality management system.
What should biotechs include in their initial RFPs to better align CDMO capabilities with project expectations?
The initial RFP should contain the entire scope of work needed from the CDMO including all ancillary activities to support manufacturing. For example, a drug sponsor needing a batch for a Phase I clinical trial might also include the following activities in the RFP: analytical method development, analytical method validation, process development, production of a demonstration batch, preparation and characterization of a reference standard, and stability studies. The RFP should be as detailed as possible and should contain information on novel raw materials or excipients. If the CDMO is in a different country from the drug sponsor, then additional details need to be addressed such as the language to be used in documentation and the currency to be used for payment. The sponsor’s wishes should be expressed in the RFP. The CDMO’s position should be clearly stated in the proposal. Drug sponsors should establish a confidentiality agreement with the CDMO before submitting the RFP. (1)
References and notes
- Ackley K, How To Select a Therapeutic Oligonucleotide CDMO, Chem Today, 2023; 41(1): 4-6.
–
Dr. Stephen Houldsworth
Sr. VP, Global Head of Small Molecules Platform, CordenPharma
Sound CDMO Considerations for Biotechs: Integrated Services, Early-Phase Technologies, Transparent Project Management & Robust Platforms
How do you align your technical team’s input when biotechs bring discovery-stage molecules with limited characterization or formulation data?
It all starts with the customer’s RFP. Our technical teams first explore the customer’s package and put together a proposal which includes core elements that we consider essential to the success of the program. These may be related to the safety of our operators, familiarization of the process, or the quality of the product and compliance with the Good Manufacturing Practices involved – for us these are non-negotiables.
We will then suggest optional elements with a business or scientific rationale as to how they will advance the asset or reduce the risk of the program, either from a manufacturing point of view or a regulatory perspective. We then present the full RFP to the customer, usually with strong suggestions regarding the optional elements which are based on our track record of experience and ability to anticipate their needs – but at the end of the day it is the customer’s decision.
In order to address limited characterization and formulation data, it helps biotechs tremendously to choose a CDMO with early-phase solid-state API and bioavailability enhancement Drug Product services to address these issues early on.
How do you plan internal resources and capacity when handling multiple biotech clients in parallel without compromising timelines or quality?
It is always a juggling act working with multiple biotech clients but our global network of Facilities, Site Project Managers, Technical Teams, Platform Directors and Sales Reps work together seamlessly to coordinate multiple biotech projects in parallel. We recommend that biotechs work with CDMOs that have large enough Process Development and Analytical resources to ensure there is always sufficient bandwidth available to accommodate shifting priorities.
How do you communicate timeline risks and mitigation strategies effectively to biotech partners?
Open communication is key, along with presenting options/solutions as challenges come up. Indicating where points of concern could impact timelines at the outset of a project is an important part of the process, as the customer may actually have valuable information they have not shared previously which can shed light on the situation or explain why they choose a particular path. These details are often crucial to informing a sound strategy.
We often present mitigation strategies up front, but since budget is an ever-present concern for the biotech customer, we usually start with the basic strategy and adjust as needed on the journey. We often find the biotech customers’ least considered aspect of planning centers around the final solid-state form of the API, and how this will impact their drug product aspirations.
Fortunately, CordenPharma has in-house Centres of Excellence for API Solid-State and Drug Product Bioavailability Enhancement considerations that address these decisions when the need arises. We encourage customers to take advantage of these services up front during development so we can maintain timelines, but we also involve the group later as challenges begin to transpire during drug product development and production.
What practices do you follow to ensure transparency, accountability, and traceability in complex development programs?
Utilizing our Smartsheet project management system, we have created a one-stop portal for all project-related details, where customers can follow along with the project progress in real time and find all project meeting minutes, action item lists, documentation trackers and of course, the all-important timeline, stored in one place.
Through this tool the customer has the most transparent access possible to their project within our systems. When multiple sites within our integrated network are involved to execute both Drug Substance and Drug Product manufacturing via our integrated supply offering, or when customers take advantage of our Centres of Excellence to solve potentially complex issues, the process can become very complicated from a project management perspective.
So here, our Global Project Management systems become extremely beneficial to biotechs by creating one common point of contact. Having that one central resource to coordinate the activities between multiple sites and priorities relieves them of a significant burden. It also creates one common vision for the project internally, which is obviously aligned with the customers’ vision.
What tools or digital platforms (e.g., dashboards, LIMS, collaborative portals) have improved real-time visibility and engagement with biotech clients?
The implementation of our common global Smartsheet Project Management system provides our customers with the most efficient access possible to our systems to track and monitor the progress of their project. Updated in real time, the system provides a common, one-stop-shop approach to project updates, project meeting minutes, timeline updates, documentation trackers, upcoming milestones and action logs. The system is obviously protected so that only the customer can securely view the project externally, but internally the full project team has access to update it as the latest information becomes available.
What’s your approach to method development and tech transfer when receiving incomplete or early-phase packages from biotechs?
When we sign on a new client, we do so on the understanding that we are here to provide whatever expert CDMO advice possible, since it’s in both parties’ interest that the program is successful. In many cases, this may be a partner’s first or second asset to be taken into the clinic, whereas in our case, we have decades of experience helping biotechs with incomplete or early-phase packages find effective method development to bring their projects through clinical phases. Some clients take advantage of that expertise, which an experienced CDMO should offer freely, and in other cases they may decide to hire CMC experts – so often a working partnership gets formed to help shepherd the asset into the clinic.
An important aspect of successful tech transfer to consider when selecting a CDMO is their facility network. Does it provide seamlessly integrated, global services that alleviate distractions and potential roadblocks to successfully meeting timelines?
–
Dr. Brett Wagner
R&D Search and Evaluation Manager, Douglas CDMO
What should biotechs include in their initial RFPs to better align CDMO capabilities with project expectations?
To ensure optimal alignment between CDMO capabilities and project expectations, biotechs should provide comprehensive and detailed information in their initial RFPs. An existing Confidential Disclosure Agreement (CDA) expedites the information exchange process, allowing the CDMO to access more granular details and formulate a precise proposal.
Biotechs should clearly define the current state of their formulation, indicating whether it’s a prototype or well-established, and specify if the CDMO is expected to undertake formulation development. If the CDMO is expected to undertake formulation development, it is very helpful that any intellectual property or patent considerations related to the formulation or process are explicitly stated to prevent future complications.
The RFP must also include a detailed project scope, clearly outlining the asset, its development phase, the desired timeline, and specific deliverables such as prototype formulation, drug product for clinical trial use, or analytical method development. A comprehensive list of all required API and finished product analytical methods, their current validation status, and any available method transfer data is also paramount. This eliminates guesswork for the CDMO and ensures they can assess their capabilities against the required analytical needs.
Additionally, finished product specifications, including parameters like color, weight, water content, and expected impurity levels, are vital for the CDMO to understand the target product profile. An example of this is that while RFPs often state the API dose, they frequently overlook specifying the desired fill weight for the final product. Providing this detail is essential, as it allows the CDMO to accurately assess manufacturing feasibility and provide realistic cost estimates for the final dosage form.
Specifying material and equipment requirements, such as preferred raw materials, excipient grades, and favored suppliers, significantly streamlines the CDMO’s proposal generation by removing assumptions.
Biotechs should also clearly indicate if regulatory assistance is required from the CDMO.
How can biotechs proactively minimize scope creep when negotiating contracts with CDMOs?
A foundational step is to define expectations and responsibilities through a highly detailed Statement of Work (SOW). The SOW should be outline every task, deliverable, and milestone, each with clear and objective acceptance criteria.
Equally important is establishing a formal change order process. This pre-agreed-upon process dictates how any modifications to the SOW will be handled, including a clear approval workflow and a mechanism for rigorously assessing the impact of proposed changes on both timelines and costs.
Regular, structured meetings with a clear agenda are also essential for proactive scope management. These frequent interactions allow both parties to track progress, identify potential deviations from the SOW early, and address them before they escalate into significant scope creep. Furthermore, explicitly defining “out-of-scope” activities within the contract prevents future misunderstandings about what is not included in the agreed-upon work.
Having a consistent project team throughout the project lifecycle, with representatives possessing appropriate authority to make decisions in the best interests of the stakeholders, is crucial. These internal representatives should be technically proficient and close enough to the project to understand its intricacies and make informed decisions.
Ultimately, a foundation of trust and good faith between the partners fosters an environment where potential scope changes can be discussed openly and managed collaboratively. Biotechs often excel in this area due to their typically small, knowledgeable, and highly invested teams, which can contribute significantly to effective scope control.
How can biotechs establish more apparent communication channels to avoid misalignment with CDMO teams?
Establishing clear and apparent communication channels is paramount for biotechs to avoid misalignment with CDMO teams and ensure project success. A key strategy is the assignment of a dedicated project manager who serves as the primary liaison between the biotech and the CDMO. This individual acts as the central hub for all communication, streamlining information flow and preventing fragmented or conflicting messages. At Douglas CDMO, we value experienced and consistent project management to fostering high customer satisfaction. Dedicated and experienced PMOs are instrumental in maintaining continuity and clarity throughout the project.
Beyond a dedicated project manager, a proactive approach to site access can significantly enhance communication and collaboration. CDMOs that welcome their customers on-site as frequently and for as long as possible foster a transparent and collaborative environment. This direct interaction allows biotech teams to engage with the CDMO’s personnel, observe processes, and address any potential issues or misunderstandings in real-time, thereby minimizing the chances of misalignment.
What steps can biotechs take to define project requirements better and avoid unnecessary delays?
Providing a comprehensive data package upfront is crucial. This includes all available data, such as process development reports, analytical data, and stability data. The more complete the initial data package, the less guesswork the CDMO needs to do, leading to more accurate proposals and efficient project initiation. Secondly, prioritizing a dedicated Q&A session during the RFP process is highly beneficial, enabling biotechs to clarify requirements, address any ambiguities, and ensure a shared understanding of the project scope.
For larger or more complex projects that require decision-making at critical points, we may suggest a phased approach. Breaking down a substantial project into smaller, more manageable phases with defined go/no-go decisions allows for incremental progress and provides opportunities to re-evaluate and refine requirements at each stage.
What should biotechs do to prepare their technology transfer packages for smoother collaboration with CDMOs?
A fundamental principle is to minimize the number of assumptions and exclusions from the outset. The CDMO’s project will likely involve multiple internal teams, and the more concrete and complete the information provided, the more efficiently they can execute. It is crucial for biotechs to have a realistic awareness of the current state of their project and its associated data. This includes ensuring that analytical methods are well-matched to the formulation for the final product. Biotechs should explicitly state upfront if their methods are ready to be transferred and, importantly, confirm that the chosen CDMO possesses the necessary capabilities and equipment to take on and implement those methodologies.
The technology transfer package itself should be a comprehensive document. This includes a detailed process description, complete raw material specifications, a comprehensive equipment list, and a list of all required analytical methods, along with their current validation status.
How can biotechs evaluate whether a CDMO’s team has adequate experience and capacity to handle multiple projects?
Biotechs should look for CDMOs with a proven track record of success, such as those that have decades of experience launching their own products into numerous global markets, like Douglas CDMO. This demonstrates not only manufacturing expertise but also a deep understanding of regulatory pathways and commercialization. During the due diligence phase, conducting a thorough site audit is essential. This allows the biotech to directly assess the CDMO’s facilities, equipment, quality systems, and overall operational capacity.
It’s also worth thinking through the supplier strategy early on. An end-to-end CDMO offering the full range of services from early development to commercialization under one roof mitigates risk, enhances efficiency, and truly accelerates time to market.
Inquiring about the experience and qualifications of the specific project team members who will be assigned to the biotech’s project is critical. The length of tenure within the CDMO’s team can be a strong indicator of stability, accumulated expertise, and a robust organizational structure.
Requesting references from other biotech clients can provide insights into the CDMO’s responsiveness, problem-solving abilities, and overall client satisfaction.
It is important to directly inquire about their experience with particular dosage forms or complex molecules that are relevant to the biotech’s project. A CDMO with specific, demonstrated experience in the required area is more likely to possess the necessary technical expertise and established processes to handle the project efficiently and successfully, even when managing multiple projects concurrently.
–
Xavier Pujol
CSO & CDMO Business Development North America, Farmhispania Group
How do you approach early-stage discussions with biotechs during RFP or feasibility phases?
A foundational step in any collaboration is ensuring that both parties are aligned scientifically and strategically. CDMOs should begin by clarifying the biotech’s needs and timelines. What activities are on the critical path? What activities are a must and what are a nice-to-have? What challenges are anticipated? Understanding these elements allows us, as a CDMO, to assess whether the project fits in our timelines and aligns with our technical capabilities and strategic focus.
Furthermore, a clear definition of scope and deliverables is essential to avoid misalignment and delays. We always work together with our partners to define what outputs are expected not only in terms of quantities and quality to be delivered but also in terms of knowledge and project understanding. Establishing clear success criteria for each project phase helps both parties meet timelines, evaluate progress and, more importantly, determine readiness for the next stage.
For us, trust is the cornerstone of any successful partnership. CDMOs should be transparent about their capabilities and limitations. If a request falls outside our core expertise, it is better to acknowledge this early while expressing a willingness to explore solutions. Sharing relevant experience and successful case studies helps us build credibility. Establishing a two-way open and frank communication has always been Farmhispania Group’s philosophy to set the tone for a collaborative and flexible working relationship.
Many early-stage biotech companies operate under tight budget constraints. We support our partners in such situations by proposing phased or modular approaches that allow for incremental investment. Flexible engagement models—such as full-time equivalent (FTE), milestone-based, or shared-risk structures—can also help align financial and technical goals. We believe that open discussions about resource availability and constraints foster realistic planning and reduce the risk of delays and extra costs.
Finally, it is important to define what happens after the initial project phase. Is there a clear plan for scale-up, IND-enabling studies, or clinical manufacturing? Understanding the biotech’s internal decision-making process, who the key stakeholders are and what triggers further investment or partnership helps us anticipate needs and plan accordingly.
At what stage in drug discovery do you recommend CDMO involvement to ensure smoother downstream development?
Involving CDMOs early in the drug development process is highly recommended, beginning with the lead optimization phase. At this stage, Farmhispania Group can offer valuable input on the synthetic feasibility, scalability, and manufacturability of lead compounds. This may help avoid selecting leads that are difficult or costly to produce, enables the early development of robust synthetic routes, and allows for the identification of potential impurities or stability issues. Such involvement becomes even more critical during preclinical candidate selection, a key decision point where CDMO insights help assess the difficulties and challenges that will need to be addressed when the chosen candidate moves into development.
During IND-enabling studies, CDMO participation is essential, as we are responsible for producing GMP-grade material, conducting stability testing, and preparing regulatory documentation. We believe that our involvement in these activities ensures a smooth transition to clinical manufacturing and reduces the risk of delays due to CMC-related issues.
What best practices help ensure scientific alignment between CDMO/CRO and biotech analytical, formulation, and process development teams?
Effective collaboration between biotech companies and CDMOs is based on early and transparent communication. This begins with kick-off meetings to align technical goals, timelines, and expectations, along with the sharing of background data on the product, prior work, and known challenges.
Defining clear success criteria for each development phase is also essential. Joint technical planning plays a key role, with both parties co-developing study designs, analytical methods, and manufacturing strategies. Joint risk assessment workshops conducted at Farmhispania Group, help identify potential scientific and technical risks, while stage-gate reviews provide structured checkpoints to evaluate data and adjust plans in a collaborative manner.
Data transparency and integration are equally important, supported as much as possible, by real-time data sharing, standardized reporting formats, and regular joint data reviews to ensure mutual understanding. At Farmhispania Group we designate technical leads and encourage direct peer-to-peer interactions among analytical chemists, process chemists and process engineers. Such cross-functional scientific teams significantly enhance the collaboration between both parties and represent a critical factor in the overall project success across its different phases.
Finally, continuous feedback and improvement are maintained through regular technical meetings, post-milestone reviews to reflect on lessons learned, and open feedback loops to refine workflows and communication practices.
How do you communicate timeline risks and mitigation strategies effectively to biotech partners?
Risks should be flagged as early as possible—well before delays become imminent—and supported with relevant data such as historical timelines, resource constraints, or technical challenges. In our case, the use of clear, visual communication tools like Gantt charts and traffic light systems help quickly visualize the project progress. Scenario planning, including best-case, expected, and worst-case timelines, further enhances clarity.
When presenting risks, we think that it’s important to include mitigation strategies. We, at Farmhispania Group, always accompany each risk by its root cause (e.g., raw material delays, analytical bottlenecks…), the potential impact on milestones or deliverables, a mitigation plan and a contingency plan in case mitigation efforts fall short.
Alignment on decision points is also critical—both teams should agree on go/no-go gates and establish clear escalation protocols for raising issues to leadership. Regular and structured weekly project updates are key for short term, tactic planning and decisions while joint steering committee meetings are essential for project strategic alignment.
How do you support “fast-to-clinic” timelines while maintaining regulatory compliance and phase-appropriate quality?
A well defined phase-appropriate development strategy is crucial to support “fast-to-clinic” timelines. This involves focusing on essential CMC elements for early-phase trials and deferring non-critical activities like full method validation and long-term stability studies to later phases.
Appropriate regulatory planning is also key. Engaging regulatory experts help define the requirements for IND submissions, avoid delays and over costs for non-critical activities. A well-structured CMC roadmap should clearly distinguish between immediate and future requirements.
A risk-based quality management approach is essential. Applying Quality by Design (QbD) principles even in early-stage projects helps start identifying critical quality attributes (CQAs) and process parameters. Risk assessments, such as FMEA, can be used to prioritize controls and testing.
Efficient supply chain management remains a critical component as well. At Farmhispania Group, we prioritize the early procurement of critical raw materials and the qualification of backup suppliers to effectively mitigate supply-related risks.
Finally, as already mentioned, transparent communication with biotech partners is vital for us. Setting realistic timelines and identifying potential bottlenecks early fosters trust and efficiency.
What qualities or behaviors define a successful biotech partner from your perspective?
In our view, a successful collaboration with a CDMO begins with an open and collaborative mindset. For us, this means being open to feedback and scientific dialogue, valuing partnership over transactional relationships, and engaging cross-functionally beyond just business development interactions.
Having a clear vision and prioritization strategy is also essential. Both parties should understand their shared goals, whether it’s filing an IND, achieving proof-of-concept, or securing a partnership. They must be able to prioritize critical path activities, make timely decisions, and weigh trade-offs between speed, cost, and risk.
Being well prepared also plays a key role. Providing complete documentation—such as compound history and analytical data—helps avoid delays.
Finally, showing flexibility and adaptability are vital in a dynamic development environment. We always strive to be able to pivot when data or circumstances change, embracing phase-appropriate and risk-based approaches together with our partners, creating solutions when standard paths don’t fit.
–
Kenneth N. Drew, Ph.D.
VP Flamma USA, Flamma
What Biotechs Don’t Know (Yet) About Getting a CDMO Proposal That Works
Engaging with early-stage biotech companies during the RFP or feasibility phase can be both exciting and challenging. These discussions are critical to the success of both parties, as they often center around a molecule still in its infancy. A well-constructed Request for Proposal (RFP) serves as a roadmap to help ensure an accurate and realistic proposal from the CDMO (Contract Development and Manufacturing Organization).
However, when working with a biotech that is new to the RFP process, challenges often arise. Inexperience can lead to a poorly crafted RFP. This is typically due to a disconnect between what the company wants and what it truly needs. This misalignment can create delays and stress already-tight timelines. Here, an experienced business development professional can provide vital guidance to help align expectations and requirements early in the process.
Building the Right RFP Foundation
For a CDMO to provide a comprehensive and accurate proposal, the biotech must supply clear, concise, and relevant information. Key items include:
• Potency of the molecule
• Required quantity (need vs. want)
• Intended use of the material
• Target timelines
• Development stage and context
The absence of this foundational data leads to flawed proposals and miscommunications. No matter how detailed the RFP, it’s critical to set realistic expectations. Process development for small molecules is inherently unpredictable. Unexpected challenges will arise. This is not due to incompetence, but because molecular behavior is often difficult to predict.
This is why I always recommend including at least a 20% contingency buffer in the budget. It’s far easier to justify having a buffer upfront than to return to a biotech’s finance team later requesting additional funds.
Expect the Unexpected
In my experience, no project has ever gone exactly as outlined in the original proposal. This isn’t unique, it’s the nature of small molecule development. Just like my Ph.D. thesis, which looked straightforward on paper but was anything but in practice, development work always brings surprises. Recognizing this from the start sets the stage for a healthier collaboration.
The goal of the CDMO is to make the biotech team look good in front of their management. That requires a true partnership built on shared risk, honest communication, and mutual commitment to the end goal: delivering high quality material quickly and efficiently to advance drug development and beat the competition to market.
The Problem with “Template” Proposals
Many CDMOs rush to generate proposals using templated pricing models that lack depth and customization. These proposals often appear attractively low-cost, and less experienced CMC professionals may mistake them as comprehensive. In contrast, our team takes a detailed, tailored approach in which we are asking the hard questions and taking the time to get the scope right. Yes, it’s more work upfront, but it ensures far fewer problems down the line.
Proposals should be sectioned and transparent. A proposal with only a total lump sum figure is essentially useless. I recommend dividing the proposal into:
- Process Development
Includes route scouting, basic scale-up chemistry, and initial analytical method development.
2. Analytical Development
Focused on phase-appropriate methods that support current and future GMP work.
3. Demo Batch Production
Typically 100 to 500 grams for early formulation or tox studies.
4. GMP Manufacturing
Ranging from 1 to 20 kg, depending on indication and stage.
5. Stability Studies
Often overlooked but essential for IND/IMPD support.
6. Safety Assessment
Evaluation of scale-up risks and thermal hazards.
Additionally, include a raw material table which is a highly effective tool to assess cost drivers, explore alternative synthetic routes, and compare CDMO proposals on a like-for-like basis. Absence of this table makes it hard to scrutinize assumptions or identify areas of cost inflation.
Avoiding the VAT and Tariff Traps
Today’s global supply chains present hidden financial risks. Some overseas CDMOs quietly pocket value-added tax (VAT) refunds instead of passing the savings back to the customer. I’ve seen this firsthand even from large, well-known firms. Be proactive. Ask how import/export taxes, tariffs, and VAT are handled. We’ve even implemented processes internally to manage VAT recovery ourselves on behalf of clients who lacked the resources thus ensuring they pay only the true cost of the work.
CDMO Partnerships Are People-Driven
Early-stage drug development is expensive, complex, and filled with uncertainty. The best way to manage this is to build trust with your CDMO. They should act as an extension of your internal team not just a vendor.
Face-to-face meetings are invaluable. Visit their facilities. Shake hands. Get a feel for the culture and people you’ll be working with. That gut feeling you get from meeting a team in person is often more accurate than any slide deck or glossy marketing campaign.
Don’t rely solely on internet ads or trade publication rankings. Those are often created by marketers, not scientists. Attend conferences like CPhI Worldwide for example, the October 2025 event in Frankfurt offers a great opportunity to visit multiple European CDMOs either side of the event.
Listen to the Wisdom of Your Network
Talk to your peers including current colleagues or trusted former coworkers. Their experiences with CDMOs are worth more than a free lunch. If you keep hearing the same CDMO name come up in conversations, take that seriously. High-quality CDMOs are often busy for a reason.
That said, be cautious of those who offer instant availability and rock-bottom pricing. In many cases, it’s a bait-and-switch tactic. They win the work with an unrealistic quote, then inflate scope mid-project when it’s too late for you to pivot.
Be Honest with Management and Yourself
Unrealistic internal timelines are a recipe for disaster. Push back when needed. Communicate the true cost of speed. A good CDMO will tell you what’s feasible and not just what you want to hear.
I’ve lost projects because I was honest about timing or budget constraints, only to have those same customers come back months later when low-cost options failed. It’s frustrating, but I’d rather be transparent than overpromise and underdeliver.
The Bottom Line
You’ll know when you’ve found the right CDMO. How? Because you took the time to:
• Develop a clear, thorough RFP
• Set realistic timelines and budgets
• Demand a transparent, itemized proposal
• Meet and evaluate the actual team you’ll work with
• Treat the CDMO as a partner, not just a vendor
The best outcomes come from partnerships based on trust, shared risk, and mutual accountability.
–
Kyriakos Kansos, George Ntortas
CDMO Consultants, Fuliginous Management Consulting (FMC)
The Evolving Interface Between Biotech Companies and CDMOs
Biotechs are at the forefront of pharmaceutical innovation, driving new therapeutic approaches ranging from monoclonal antibodies to mRNA, cell, and gene therapies. But bringing these breakthroughs from lab bench to patients is an intricate journey that often depends on robust, strategic partnerships with CDMOs.
The collaboration between CDMOs and Biotech companies differs from that with traditional pharmaceutical companies (mainly marketing small molecules), reflecting each partner’s structure, needs, and development stage. Biotechs typically engage CDMOs early, often from lead optimization or preclinical phases, seeking scientific guidance, formulation support, and regulatory preparation. These partnerships are agile, informal, and heavily reliant on CDMO’s technical expertise, as Biotechs often lack internal CMC resources. CDMOs act as strategic partners, filling data gaps and accelerating timelines toward first-in-human trials. In contrast, traditional pharma companies tend to engage CDMOs for capacity expansion, specialized technologies, or cost-effective commercial supply. Their projects are usually better characterized, governed through formal processes, and supported by in-house CMC expertise. While Biotech-CDMO collaborations are defined by scientific innovation and flexibility, traditional pharma-CDMO partnerships emphasize operational rigor and long-term scalability. As CDMOs evolve, they offer integrated early-development platforms for biotech clients adapting their governance, capabilities, and culture accordingly.
Understanding how to build and maintain an effective interface between biotech companies and CDMOs is vital for accelerating timelines, ensuring product quality, and enabling long-term commercial success.
Early-Stage Collaboration: R&D and Process Development
Biotech innovations typically emerge from a research-driven mindset focused on mechanism of action, proof of concept, and early clinical results. CDMOs support in high-throughput screening, molecular biology, cell line and vector development, in vitro and in vivo studies. Additionally, CDMOs bring a different but complementary focus: robust process engineering capabilities, scalability, quality systems, technology platforms, and regulatory compliance that enable efficient scale-up.
For a successful and mutually beneficial partnership, alignment begins during the early development phase. Even before a clinical candidate is selected, discussions about manufacturability, formulation feasibility, and process development should be underway. This collaboration ensures that development strategies anticipate regulatory expectations, potential scale-up constraints and reduce the risk of costly redevelopment further down the line.
Clinical Trial Manufacturing: The Critical Middle Ground
The manufacturing of clinical trial material is a pivotal but often underestimated phase in the biotech product lifecycle. CDMO’s role shifts from technical collaborator to operational executor, managing GMP production, labeling, packaging, and sometimes even clinical supply distribution.
Clinical trial manufacturing introduces several layers of complexity:
• Production must occur under strict timelines tied to trial start dates.
• Yields may be low and processes still under optimization.
• Regulatory documentation must be developed in parallel with ongoing studies.
For early-stage biotech companies, delays at this stage can be existential. A well-integrated CDMO mitigates this risk by offering flexible capacity, small-scale GMP suites, and adaptive planning tools.
Tech Transfer: Turning Science into Process
Tech transfer is a multifaceted process that translates lab-scale protocols into industrial production, whether for clinical or commercial use. For biologics and advanced modalities, this often involves transferring not just methods but knowledge: rationale behind process conditions, critical quality attributes, and material behavior.
Challenges frequently arise from incomplete documentation, different equipment footprints, or unaligned analytical methods. These risks can be mitigated by:
• Engaging in structured tech transfer planning
• Assigning cross-functional leads from both sides
• Using a defined acceptance criteria checklist
Tech transfer success depends on clarity, consistency, and collaboration not only between teams but also between technical philosophies.
Supply Chain Integration: Beyond Manufacturing
A common misconception is that CDMO collaboration ends with batch production. In reality, supply chain integration is equally essential, especially for biotech companies developing global clinical or commercial programs.
Key supply chain concerns at the biotech-CDMO interface include:
• Lead times and raw material security
• Cold chain and temperature-controlled logistics
• Forecasting accuracy and capacity alignment
• Inventory management and regulatory labeling
Miscommunication in these areas can lead to stockouts, compliance issues, or financial penalties. To prevent such issues, successful partnerships build integrated planning cycles that include demand review, scenario planning, and real-time visibility dashboards.
Regulatory Interface: Coordinated Compliance
Regulatory approval is the culmination of years of development but it also requires meticulous preparation. CDMOs are deeply involved in the preparation of Chemistry, Manufacturing, and Controls (CMC) sections of regulatory filings, as well as in responding to agency queries.
A strong interface must cover:
• Early alignment on regulatory expectations (e.g., ICH guidelines, FDA/EMA requirements)
• Coordination of documentation timelines
• Clarity on roles for GMP inspections and audits
• Unified narratives for product comparability and process changes
Regulatory misalignment can cause months of delay. Early-stage planning and open communication throughout development mitigate these risks significantly.
Building the Right Interface Model
The Biotech-CDMO collaboration model is shifting from a transactional approach (outsourcing execution while retaining all strategy) to co-development of products and investing in strategic partnerships with shared risk.
Successful Biotech-CDMO interface typically shares three characteristics:
1. Cross-Functional Engagement: Involving QA, Regulatory, Project Management, and Supply Chain—not just R&D.
2. Governance Structure: Regular joint steering committees, issue escalation paths, and KPIs.
3. Cultural Fit: Shared values around transparency, accountability, and problem-solving.
The Future of CDMO-Biotech Collaboration
Looking ahead, the lines between Biotechs and CDMOs will continue to blur. We would see more co-development models, risk-sharing agreements, and even equity-based partnerships. Digital transformation—through data integration, advanced analytics, and AI-driven process optimization—will further enable real-time collaboration and innovation.
Ultimately, the ongoing evolution of Biotech-CDMO partnerships is reshaping the pharmaceutical industry for the better. By combining scientific ingenuity with operational excellence, these collaborations promise to deliver new therapies to patients more efficiently and reliably than ever before.
Conclusion
As biotech products grow in complexity, so must the partnerships that support them. CDMOs are no longer just service providers—they are co-navigators in the journey from innovation to commercialization. Strengthening the interface across technical, operational, and regulatory areas is essential for ensuring that breakthrough therapies reach the patients who need them, faster and more reliably.
For both Biotech companies and CDMOs, the future lies in collaboration—not as a reactive necessity, but as a proactive strategic asset.
–
Nicholas Shackley
CEO, Gannet BioChem
What are the top three criteria when selecting a CDMO for complex drug modalities like ADCs or oligonucleotides?
When selecting a CDMO (Contract Development and Manufacturing Organization) for complex modalities such as antibody-drug conjugates (ADCs) or oligonucleotides, there are three foundational criteria that companies should carefully assess: expertise, cultural fit, and scalability.
Primarily, a CDMO’s track record of technical expertise in both early development and late-stage commercialization is essential. These processes are highly complex and demand a nuanced understanding of not only chemical synthesis and purification but also specialized technologies, such as linker-payload chemistry for ADCs or solid- and solution-phase synthesis for oligonucleotides. A history of successfully advancing multiple programs through IND, NDA, and commercial launch phases—across regulatory jurisdictions—offers strong confidence in capability and reliability. Whenever possible, teams should review case histories or request anonymized examples of similar projects the CDMO has completed, especially for programs with comparable risk profiles or timelines.
Second, cultural alignment and partnership mindset are often underestimated but are crucial for long-term project success. Even the most technically capable CDMO may fall short if there is a mismatch in communication style, escalation pathways, or prioritization practices. Biotechs should assess whether the CDMO views them as a strategic partner rather than just another client in the queue. Responsiveness, transparency, and the ability to adapt across time zones or regions contribute to operational efficiency and trust. Proximity also matters—while not always necessary, geographic closeness can reduce communication delays, facilitate site visits, and support real-time collaboration during critical phases.
Third, the ability to scale—in terms of both human capital and infrastructure—is essential to avoid bottlenecks as the program transitions from clinical to commercial. This includes having sufficient analytical, formulation, and kilo lab capacity, along with a qualified team of scientists, engineers, and regulatory experts to support rapid iteration and issue resolution. CDMOs that proactively invest in expanding capacity ahead of demand demonstrate strategic foresight and reduce the risk of a project being deprioritized or delayed due to resource constraints.
How vital is a CDMO’s track record of on-time project completion, and what evidence should be reviewed?
Timely execution is one of the most critical metrics in a CDMO partnership—especially in an environment where biotech funding, competitive positioning, and regulatory milestones are tightly coupled to development timelines. A CDMO’s history of meeting deadlines is not just a project management issue—it reflects the organization’s ability to coordinate cross-functional teams, anticipate risks, and execute reliably under pressure.
To assess this, biotechs should request project case studies that illustrate how the CDMO has handled challenges such as material delays, analytical troubleshooting, or regulatory changes. These case studies should outline not only what went wrong, but also how the CDMO communicated, adapted, and resolved the issue. Authenticity is critical—if the examples feel overly polished or lack detail, it may be a sign that the CDMO lacks experience with complex problem-solving in real-world scenarios.
Additionally, market intelligence plays a crucial role. Informal references from peers, industry consultants, or even past clients can reveal insights not captured in formal materials. Feedback on responsiveness, escalation handling, and the ability to stay on schedule provides a more comprehensive picture of the CDMO’s operational maturity.
What red flags might indicate a CDMO could struggle with on-time delivery?
Several early indicators can help companies identify potential risk areas before they manifest into missed milestones.
One red flag is the speed and quality of communication during the initial phases—specifically the CDA (Confidential Disclosure Agreement), RFP (Request for Proposal), and quotation process. If timelines slip during these early steps, or if information is inconsistent or incomplete, it may signal deeper organizational inefficiencies.
Another concern is limited flexibility in resource deployment. Some CDMOs operate with a fixed capacity model where assets and headcount are tightly allocated across programs. This rigidity can become a significant problem when unexpected challenges arise and additional support is needed. In such environments, the “risk/time/cost” equation becomes heavily biased toward cost, as the CDMO cannot deploy supplemental resources without disrupting other programs.
Finally, project prioritization within the CDMO’s broader portfolio matters. If the organization is heavily committed to large commercial supply contracts, your development-stage program may struggle to get adequate attention—especially in QA and QC functions that are typically capacity-constrained.
What indicators demonstrate a CDMO’s expertise in handling complex programs such as ADC conjugation or oligonucleotide synthesis?
Biotechs should look beyond marketing materials and assess hard indicators of modality-specific experience. This includes the number and nature of ADC or oligo programs handled to date, the stages of the lifecycle those programs were in, and the regulatory jurisdictions supported.
Equally important is the CDMO’s investment in infrastructure and talent. Have they built conjugation suites with appropriate containment? Are oligonucleotide synthesizers in place to support both early development and scale-up? Is there a bench of subject matter experts (SMEs) who understand payload stability, impurity control, and analytical method development?
These investments should also demonstrate alignment with the speed and complexity of modern drug development. It’s one thing to have capabilities on paper—it’s another to show that these resources are accessible, integrated, and purpose-built to accelerate programs through critical milestones.
How should companies evaluate a CDMO’s quality and infrastructure management system during due diligence?
A comprehensive evaluation of the Quality Management System (QMS) is central to any CDMO partnership. This typically begins with an on-site audit, which should include a review of SOPs, batch records, deviation and CAPA (Corrective and Preventive Action) logs, and change control processes. The goal is to ensure that the QMS is both compliant and adaptable—able to support phase-appropriate development while being rigorous enough to pass regulatory scrutiny.
Biotechs should also assess the CDMO’s regulatory compliance history, including inspection outcomes from agencies such as the FDA, EMA, or MHRA. Transparency around findings and their resolution history is essential. If possible, companies should request summaries of recent audits, review how quickly and thoroughly responses were handled, and gauge the CDMO’s attitude toward continuous improvement.
From an infrastructure perspective, physical site capabilities must match the demands of the program. This includes evaluating available equipment, HVAC systems, containment strategies, and expansion plans. A CDMO that is open about its near-term capital plans and can demonstrate agility in closing infrastructure gaps is far more likely to be a reliable partner in a dynamic development environment.
How can biotechs ensure that CDMOs meet agreed timelines without compromising quality?
Meeting timelines without sacrificing quality requires strong alignment, open communication, and shared accountability from the outset. Establishing a detailed project plan—with agreed-upon deliverables, risk triggers, and escalation pathways—sets the foundation. Both sides should review interdependencies and proactively plan for resource requirements at critical points in the program.
A useful approach is to build in buffer capacity or “over-scope” in areas like yield assumptions or analytical throughput. This allows the program to absorb minor deviations without triggering timeline delays. Having flexible access to surge resources, particularly in analytical and QA areas, is often more effective than relying solely on punitive clauses.
Financial incentives and penalties can be structured into the agreement but are rarely sufficient on their own. The most sustainable strategy is to cultivate a partnership mindset, where the CDMO shares ownership of the outcome and is empowered to act decisively in the face of challenges.
How should organizations evaluate the realism of CDMO timelines—especially in fast-to-clinic scenarios—and prepare for potential delays?
In accelerated programs, timeline credibility is paramount. Companies should benchmark proposed timelines against internal data and use external consultants when needed to validate assumptions. It’s essential to review whether the CDMO has adequately resourced the project and whether those resources are booked in advance or contingent on availability.
Conducting a structured risk assessment helps identify where slippages are most likely to occur and allows contingency measures to be embedded into the project plan. This should extend across the full value chain—including development, manufacturing, testing, release, and fill/finish—to avoid surprises later.
To prepare for delays, companies should ensure that all stakeholders—from program management to clinical operations—understand the impact of each step on the overall timeline. Pre-booking critical slots such as fill/finish or stability studies, and ensuring downstream flexibility, can mitigate cascading effects from upstream delays.
Ultimately, the best safeguard is early alignment on risk, realistic resourcing, and clear accountability mechanisms to keep programs on track without compromising quality.
–
Robert Hughes
Research Fellow, Grace
The Secret Ingredient for a Successful CDMO/Biotech Partnership: Communication
How do you communicate timeline risks and mitigation strategies effectively to biotech partners?
We strive for transparency, proactive communication, and a solution-oriented approach when communicating timeline risks and mitigation strategies with our biotech partners.
The program manager is foundational to this – they act as the main point of contact on our side and help facilitate communications between the right parties from both companies.
During the quotation and pre-IND stage, an initial risk assessment presents a high-level overview of potential risks to the API process and timeline. Risk factors include the complexity of the synthetic route, availability of starting materials, impurity profiles, and analytical method development. Mitigation strategies should be outlined, which may include alternative synthetic routes, dual sourcing of starting materials, early impurity profiling, solid form screening, and an analytical method development plan. This is also the time to present a preliminary timeline along with built-in buffers – we know that there will inevitably need to be adjustments along the way, so we try to account for this up front.
As the project progresses to IND-enabling studies, maintaining a detailed risk register that lists all identified risks, their probability of occurrence, potential impact on the timeline, and proposed mitigation strategies is essential. Critical path analysis should identify the longest sequence of tasks that will determine the overall project duration. Communication frequency should be adjusted based on the project stage, with more frequent updates during critical phases.
Once the project moves into clinical studies, communicating potential disruptions to the API supply chain, as well as progress on process and analytical development and eventually process validation activities, is essential. Impurities continue to be monitored, along with opportunities to optimize the process to improve yield, reduce costs or enhance quality.
A dedicated program team at the CDMO, composed of experts from various fields such as analytical services, process R&D, engineering/operations, and quality, is critical for providing comprehensive communications, including technical guidance and regulatory support. Regular and flexible meetings keep clients informed of progress, with the program manager acting as the single point of contact. By fostering strong relationships, maintaining consistent team members, and facilitating direct interactions with technical experts, CDMOs can help biotech companies navigate the complexities of drug development.
How do you support “fast-to-clinic” timelines while maintaining regulatory compliance and phase-appropriate quality?
I can’t stress enough the value of ongoing communication at regular intervals throughout the entire process to align expectations, priorities and timelines. This goes for both internal teams (process R&D, analytical services, quality) as well as external communications with the sponsor.
It starts with having a thorough understanding of the sponsor’s expectations from the quotation stage. This ensures we can meet their needs and identify any deficiencies in the technology package, such as final form selection, analytical gaps, and state of the synthetic route to the API. We recognize that not all sponsors have the same level of understanding of the requirements for transitioning a program from pre-IND to clinical stages. In some cases, we may recommend hiring a consultant to help bridge this gap.
The goal is to optimize the process to realize a balance between speed/efficiency and quality/compliance. To achieve this, consider the following:
• Build an extensive collection of data from experiments, utilizing DoE and machine learning, to build the process knowledge necessary for supporting “fast-to-clinic” timelines.
• Aim for a scalable process that is representative enough for tox lot production, with fine-tuning occurring in preparation for phase 1 production.
• Understand impurities, even at early clinical development stages, including their origin, formation, and control mechanisms.
• Conduct activities in parallel whenever possible to shorten the overall timeline. For example, analytical method development should occur alongside process development activities on the synthetic route to the API.
• Secure a supply chain for raw materials, or partner with a CDMO that can self-supply or connect you to their network of verified suppliers.
• Employ risk-based decision making, prioritizing activities based on their potential impact on safety and efficacy.
Supporting an expedited timeline means simplifying some processes and focusing on essential activities, which might include analytical testing that is appropriate for IND and initial clinical trials. At the pre-IND stage, this might look like prioritizing a viable synthetic route, even if it is not fully optimized, or identifying a preliminary solid form with acceptable properties, rather than the perfect form for now.
Another key component is utilizing the kilo lab as a critical intermediate stage to assess the manufacturing process and address any issues before moving into the next stage of production. Partnering with a CDMO that has designed their kilo lab to mirror large-scale production can potentially save you time and money in the long run.
A solid understanding of the sponsor’s expectations, combined with effective communication, process prioritization, and an emphasis on safety and efficacy, enable our team to support a sponsor’s “fast to clinic” timelines.
–
Kit Hale, Ph.D.
Founder & Principal, Hale CMC Solutions
What should biotechs include in their initial RFPs to better align CDMO capabilities with project expectations?
A properly-scoped project is the first step in developing a strong collaboration between a biotech and a CDMO partner. As the sponsors of new projects, the initial responsibility lies with the biotech to properly define what activities they need a CDMO partner to perform. The type of information included in a thorough request-for-proposal (RFP) depends on the stage of development and might often include company & project background, deliverables and timeline, materials, technical objectives, process descriptions & batch history, specifications, documentation requirements, and communication requirements.
Background
A brief background is often provided describing the stage of development of the asset, the anticipated clinical indication, and some information about the sponsor’s mission and corporate objectives. The purpose of this section is to introduce your biotech to the CDMO and give a high level summary of the asset. A summary of previous production batches (both non-clinical and clinical) may be included which provides additional high-level information on previous batch sizes and the overall state of the project.
Deliverables and Timeline
The key project deliverables and requested timeline should be described. This is often the most critical section of the RFP, as it provides specific information regarding how much product the CDMO needs to manufacture, the required timeline for delivery, and the necessary GMP-compliance status (whether non-GMP or cGMP). These deliverables are often linked (internally) to the sponsor’s key corporate goals such as initiating a clinical trial or filing an IND or NDA. Additional deliverables often include items like stability studies, analytical methods verification/validation, or project-specific process development or analytical activities. Regarding the project timeline, it is important for the biotech to understand that the completion of manufacturing is usually not the date at which a batch of drug substance (DS) or drug product (DP) can be shipped. For example, QC testing of a cGMP batch of clinical-stage DS or DP can often take 2-4 weeks, and based on the batch record review process, it may take another 2-4 weeks by the sponsor’s quality unit to appropriately disposition the batch and allow for shipment of fully-released material.
Materials
Information on what materials (ie regulatory starting materials, cGMP intermediates, drug substance, bulk tablets or capsules) the sponsor will provide and what the CDMO needs to source (ie raw materials and reagents, excipients, packaging materials) should be clearly described. The biotech should describe the quantities of materials they will supply and the anticipated timeline for their availability at the CDMO site. Moreover, if any analytical markers (ie impurity markers) or reference standards will be provided, these can be included in this section.
Technical Objectives
Technical objectives are often quite different between early-phase clinical projects and late-phase or commercial projects. For early to mid-phase programs, heavy emphasis may be placed on developing an initial process capable of producing the first few kilograms of DS or DP, general process and analytical development, increasing yield and purity, improving cost of goods, and decreasing process-mass intensity (PMI). If there are any aspects critical for technical development, such as mutagenic impurity control strategies or studies to ensure final polymorphic form of drug substance, these should be described. For later stage programs, more emphasis may be placed on ensuring process robustness, design-of-experiments (DOE) activities, preparing for process performance qualification (PPQ), and improving cycle times.
Process Descriptions & Batch History
Process descriptions and batch history play a key role in helping the CDMO understand the state of the process and how much time and effort are required to achieve the deliverables. The information provided here should be a complete process description for each step of the manufacturing process, as well as the batch size that each step has been performed on. If any specialized equipment is needed, it should be described. A summary of the available analytical methods should be included, so the CDMO knows what, if any, new methods need to be developed. Lack of sufficient information in this section may lead to the CDMO over- or under-bidding the project resulting in either change-orders or wasted budget, and in some cases may result in manufacturing timeline delays.
Specifications
Regarding specifications, the desired specification of the drug substance or drug product should be disclosed. For drug substance RFPs, providing specifications for intermediates and starting materials are also useful, especially if there is an existing process in-place, for example at a primary manufacturing site. Test methods and limits should be shared so the CDMO understands the quality attributes that need to be achieved.
Documentation & Communication
Documentation and communication requirements from the biotech should be outlined. These should include what documents the biotech expects to review and approve (ie, master batch records, executed batch records, deviation reports, change controls, specifications, analytical monographs, method validation protocols and reports, etc…). It is often helpful to provide the timeline that the biotech needs to review each type of document (ie 5 days for an MBR), as this information will feed into the CDMOs project timeline planning. On communication cadence, the biotech should describe how often and what types of updates they expect to receive, such as weekly written technical updates, analytical development progress, teleconference cadence, development summaries, and campaign summaries.
Additionally, regarding overall proposal expectations, pre-project timeline and budget requirements are useful to communicate with the CDMO early-on in discussions. Clearly stating milestones between RFP submission and project kick-off are often helpful to maintain timelines during pre-project discussions and negotiation. These milestones may include timeline expectations for initial RFP questions, deadlines for receipt of a proposal, the anticipated proposal acceptance target, and the desired project kick-off date. Requests for budget details may also be made (ie itemization of costs associated with analytical & manufacturing activities, raw material procurement, & waste disposal). Identification of pre-project milestones and budget requests are especially useful when RFPs are being distributed to several CDMOs for evaluation to keep multiple parties on a similar schedule and for easy proposal comparison.
With the RFP in-hand, it should be very clear to the CDMO what the requirements are in terms of deliverables as well as whether or not the CDMO will be a good technical fit considering their manufacturing infrastructure.
What should biotechs do to prepare their technology transfer packages for smoother collaboration with CDMOs?
After the RFP is submitted and an acceptable proposal is received from CDMO, the next steps are awarding the project and preparing the technology transfer package (TTP). Technology transfer is the transfer of existing processes and methods from the biotech to a new CDMO partner, and a carefully prepared TTP will set the project up for success from the onset. A well-prepared TTP will include as much information as available relevant to reproducing the process and analytical methods. Obviously, for early-stage projects or initial cGMP batches for Phase 1 studies, the available data in the TTP may be quite limited, but by later-stage development, a more robust data package is typically available.
For each manufacturing step, the most current set of process instructions and a process flow diagram are helpful, along with batch history, typical analytical data for in-process checks, and typical data for intermediate or final product release analysis should be provided. Current analytical methods should be shared to aid in method transfer. In addition to these details, any available information on process hazards, material incompatibilities, and cleaning solvents are useful. Finally, a summary of key process changes for each step throughout the development lifecycle is instructive.
In addition to a written TTP, it is advisable for the biotech to also share representative samples of starting materials, intermediates, drug substance and/or drug product with the CDMO. This will help the CDMO better understand the material properties and have a direct in-house comparison of key attributes between historical batches and present batches. The availability of historical batches is also helpful to evaluate the success of methods transfer and aid in development in case method modifications are needed. Overall, the availability of representative samples will help to ensure the technology transfer was successful and enable the CDMO to generate materials of comparable quality.
How important is it for biotechs to have an internal project manager to oversee CDMO interactions?
It depends on the stage of development, project scope, and technical team’s experience working directly with CDMOs. For example, an experienced drug substance team consisting of a process chemist, an analytical chemist, and a quality assurance expert supporting a single batch of DS intended for a Phase 1 study may not need a project manager to keep the project on track. However, as development progresses, manufacturing timelines between DS and DP, and their impact on regulatory submission timelines and clinical trial initiation quickly begin to overlap and become more complex. A project manager in this situation would be highly beneficial to maintain a holistic view of the program timeline across multiple CDMOs and parallel or dependent activities.
What collaborative approaches have worked for biotechs to build trust and accountability with their CDMOs?
Building trust and accountability between biotechs and CDMOs is a process which takes time and commitment from both sides. The most important element of this is routine honest communication across all parts of the collaboration ranging from interactions at the business-development level all the way down to discussions of technical details. On business development discussions, building trust means being honest and transparent about order projections and potential uncertainties due to clinical or toxicology read-outs. This also means obtaining efficient approvals for SOWs and POs and ensuring invoices are paid expeditiously.
In collaborations with technical teams, I like to foster a relationship focused around understanding challenges and troubleshooting together. There is a critical link between the CDMO’s technical team working on the chemistry every day and the biotech’s technical team who needs to understand the challenges the CDMO is facing and provide guidance and support. This means the biotech should recognize and understand that problems and challenges are very common during development, rather than taking an adversarial approach. Additionally, the biotech should show an active and engaged interest in the CDMOs activities. This means reviewing and providing timely feedback on written reports and coming to teleconferences with topics and questions for discussion. If possible, a technical visit to the CDMO’s R&D and/or manufacturing site in-person is highly beneficial. This level of commitment and investment from the biotech will demonstrate to the CDMO the importance of the project, keep objectives aligned, and help bridge cultural and geographic barriers in the global biotech-CDMO network.
–
Eduardo Paredes, Ph.D., M.B.A.
SVP of CMC, Leal Therapeutics, Inc.
Beyond Execution: Redefining Scientific Partnerships Between Sponsors and CRDMOs
The successful management of the Sponsor:CDMO relationship has evolved in the last few decades. This challenge of how CMOs and sponsors develop and assess scientific depth and team experience (Sponsor Q3 and CDMO Q14) has been debated since the emergence of outsourcing in the 1970s. The outsourcing of expertise—whether for compliance, process optimization, or emerging technologies—has driven the steady growth and evolution of CMOs. Increasing pressure to do more, know more, and deliver greater value has pushed CMOs beyond their original role of process execution. By the turn of the century, as they took on additional responsibilities, CMOs evolved into CRDMOs by incorporating Development and, increasingly, Research capabilities.
Sponsors today often engage CRDMOs not only for process execution, but also to collaborate on scientific research proposals that demonstrate technical depth and may introduce strategies the sponsor had not yet considered. This collaborative scoping phase allows sponsors to assess both the CRDMO’s capabilities and its potential for innovation. To prepare for this, CRDMOs frequently invest in internal R&D or platform technologies that inform their proposals and readiness for novel modalities. However, these investments can be costly and carry limited near-term return, as the specialized expertise may initially apply to only a single sponsor’s program. CRDMOs must therefore strike a balance—selectively advancing new technologies while maintaining focus on core capabilities that sustain their broader portfolio.
Meanwhile, sponsors—particularly emerging Biotechs—operate under immense pressure to progress programs rapidly with limited internal resources. Managing complex modalities like oligonucleotides, peptides or drug conjugates often involves coordinating multiple CDMOs across synthesis, conjugation, formulation, and analytical workstreams, which increases operational complexity and misalignment risk. To reduce this burden, sponsors frequently seek to centralize development within a single, technically integrated CRDMO. This “one-stop shop” model is increasingly valued in the marketplace and can become a strategic differentiator for CRDMOs looking to stay at the forefront. Yet while this model offers near-term efficiencies for the Biotech, it can create longer-term risks; overreliance on a single vendor may necessitate diversification later in development to mitigate supply chain vulnerabilities or protect intellectual capital.
This diversification may involve engaging other external CDMOs or building internal capabilities, which can introduce tension into the sponsor–CRDMO relationship. Transferring processes midstream or before the CRDMO can achieve a return on the investment may dilute a CRDMO’s incentive to invest in bespoke expertise, and spreading know-how across multiple vendors can weaken trust and continuity. One way to manage this tension is through structured, phased partnership models—where a lead CRDMO supports development through IND or Phase 1, with built-in provisions (e.g., right of first refusal, milestone-linked transitions, or defined tech transfer frameworks) for knowledge consolidation and risk-sharing as the program evolves. This approach promotes scientific continuity in early phases while preserving flexibility for later-stage decisions.
Another, often underappreciated, challenge in this relationship is its human foundation. Continuity in scientific relationships is critical to long-term success. Movement of scientific talent from CRDMOs to sponsors is common, as sponsors seek individuals with hands-on development experience and cross-modality execution skills. Conversely, CDMOs tend to recruit sponsor-side scientists but primarily into senior roles to better align with client expectations and improve translational strategy—creating a successful, dynamic but uneven talent exchange. Ultimately, the success of the sponsor–CRDMO relationship depends on sustained investment in training and mentorship within CRDMO teams. When experienced leaders continuously develop junior staff, CRDMOs can maintain a robust and flexible workforce that is resilient to attrition and capable of executing an optimized portfolio and meeting sponsor expectations.
In conclusion, the interplay between scientific depth, operational capability, and team experience remains central to the evolving sponsor–CRDMO partnership. The most successful relationships are built on early-stage collaboration, clear incentives, and shared investment in both innovation and people. By adopting structured engagement models and committing to talent development, both sponsors and CRDMOs can achieve greater alignment, reduce risk, and accelerate the path from discovery to development.
–
Abdel Aziz Toumi
CEO, Lupin Manufacturing Solutions
How CDMOs and Biotechs Can Collaborate for Successful Drug Discovery
Collaboration between biotech companies and CDMOs is becoming a key enabler of drug discovery & development. With growing focus on complex modalities like ADCs, Peptides Oligonucleotides etc., biotech companies are increasingly reliant on CDMOs. And they need a robust framework in selecting the right partner as the partnership holds the key to progress.
In this article, we present a framework that a Biotech company can employ in choosing a CDMO. We discuss the key questions that can serve as a starting point to building a comprehensive assessment of a CDMO. We also talk about aspects that a Biotech company needs to be mindful of to ensure success in the partnership.
Choosing a CDMO partner who CARES
Cultural fit, Agility, Regulatory Record, Expertise and Scalable Infra are the key parameters to be evaluated by a biotech company while choosing a CDMO partner
Cultural Fit
Cultural fit is more often an underestimated factor in evaluating a partnership. A good cultural fit inspires confidence in each other to achieve success. It encourages doing the right things, promotes transparency, proactively address challenges and resolve conflicts.
To evaluate this, the Biotech company needs to be curious, ask thoughtful questions and listen to each other. It is important for the CDMO to display the same.
For instance, Western cultures tend to be more open and direct while in the East, there is deference to hierarchy, and it is considered rude to be direct. Being aware of this nuance can help setting the right expectations from the partnership.
Agile CDMO
Progress is not always linear, and it is important to have a CDMO who can respond with agility.
Is the CDMO able to foresee challenges?
How do they embed risk mitigation actions in their project plan?
How does the CDMO structure its delivery team?
It is important for the CDMO to assign a project manager with relevant biotech experience who can streamline communication, predict risks and push for results.
A CDMO that runs discussions with detailed project plans, providing visibility into resource utilization, and collaboratively works with the Biotech to anchor timelines to reality are indicators that the CDMO can deliver on timelines, and act with agility.
Regulatory compliance
Having a clean regulatory record with agencies like FDA, EMA, ANVISA etc is testament to how the CDMO’s attitude towards quality and regulatory compliance. Non-compliance can be detrimental and a single warning letter could seal the fate of the project.
A Biotech company can look at SOPs, audit reports, deviation and CAPA management and assess data integrity practices of the CDMO.
Expertise & Scalable Infra
When dealing with complex modalities, having the right people who have proven expertise is critical for progress. For a biotech company, a CDMO with scalable infra means a one stop shop from clinical to commercial scale.
In assessing this, a biotech company needs to ask the following questions:
Does the CDMO have the right talent and the right infra?
Is there frequent churn in their teams?
What is the level of the CDMO’s understanding of the equipment needed, regulatory requirements etc.?
How soon is the CDMO able to allocate resources to your project?
Are there aspects that the CDMO needs to outsource?
How can Biotech companies contribute to a successful partnership?
As in any partnership, the goals are shared, and so are the responsibilities.
Start with the scope – A well defined scope ensures clarity, avoids project overruns and focusses energy on delivery. It is important to invest time in clearly defining the scope right from the RFP stage. This can later be enhanced based on ensuing discussions with the CDMO.
What are the deliverables?
What are the milestones?
What are the regulatory requirements?
What is the volume forecast?
It is also recommended to list down what is not needed. Based on discussions, the parties can incorporate risks and mitigation plans.
Define KPIs:
• OTIF
• % Issues notified within timelines
• % Issues resolved within agreed timelines
• % Overdue deviations
• Adherence to documentation submission timelines
• Lot Acceptance Rate
• CSAT score
Focus on Tech Transfer – Executing a successful tech transfer is crucial to ensure safe and efficient production of medicines, accelerating time to market.
• Put together a team – Set up a project team drawing from R&D, Manufacturing, Quality and Regulatory Affairs. Ensure regular check-ins.
• Scope & Risk assessment – Define scope & timelines. List all the equipment, material, process details, product details and analytical methods. Perform a gap analysis to identify gaps and develop mitigation plans accordingly.
• Maintain and share a comprehensive set of documentation – Detailed documentation covering product reports, process reports, analytical methods, validation reports, equipment specifications and quality control data to be shared with the CDMO. This is crucial as it enables the CDMO to replicate the process.
Project Manager is the key – Clearly defining the Project Management structure with a robust governance mechanism brings clarity on responsibilities and avoids siloed conversations.
For instance, without a Project Manager to streamline requirements, additional requirements can creep in from many sources and the CDMO delivery team could get distracted, leading to delays.
A robust governance structure enables transparent communication, smooth decision making and build mitigation plans for potential risks.
A robust governance structure is anchored by
1. Identifying the right stakeholders and defining responsibilities & accountabilities
2. Defining forums and their agenda
3. Project Managers of the Biotech and the CDMO as the owners of the governance structure
–
Saharsh Davuluri
Vice Chairman & Managing Director, Neuland Laboratories Limited
What best practices help ensure scientific alignment between CDMO/CRO and biotech analytical, formulation, and process development teams?
Early alignment of scientific objectives and methodologies is essential to ensure a unified approach. Structured governance forums facilitate cross-functional collaboration among R&D, Analytical Services, and Quality Assurance teams.
As a drug substance manufacturer, we utilize a robust, digital project management platform that serves as a single source of truth—offering real-time visibility into project milestones, and proactive risk identification and mitigation plans. Through dynamic dashboards, Gantt charts, and performance metrics, we ensure proactive decision-making and cross-functional alignment. Our structured governance forums—both internal and customer-facing—enhance transparency, accelerate decision-making, and ensure alignment on scope and timelines.
Comprehensive documentation protocols embedded in our workflows ensure full auditability, traceability, and compliance with global regulatory and quality standards throughout the project lifecycle.
As the asset advances through development, relevant sections of ICH guidelines and regulatory directives are systematically integrated. This includes establishing specifications, controlling impurities, setting residual solvent and nitrosamine (NDSRI) limits, and defining impurity profiles. Analytical methods are developed to detect these parameters, incorporating customer feedback to ensure the drug substance is processed appropriately, and final API (active pharmaceutical ingredient) releasing methods are aligned with drug product CMDOs.
Technology transfer activities are initiated in parallel with late-stage development, ensuring that process knowledge, analytical methods, and quality standards are effectively communicated to manufacturing sites. This enables seamless scale-up, consistent product quality, and regulatory compliance during commercial production.
How do you plan internal resources and capacity when handling multiple biotech clients in parallel without compromising timelines or quality?
CDMOs face the challenge of supporting several biotech clients in parallel while maintaining timely deliveries within the agreed quality parameters. Clear communication, agility, proactive risk identification and a culture of continuous improvement is essential for effective planning.
a) Accurate Demand Forecasting
The organization utilizes client input and historical data to anticipate resource requirements. These forecasts are updated regularly to reflect changing needs and ensure preparedness.
b) Resource Mapping and Analysis
Staff, equipment, and facilities are inventoried systematically to identify gaps. This analysis informs decisions related to hiring and infrastructure expansion.
c) Flexible Resource Allocation
Teams are cross-trained, staggered shifts are implemented, and contingency staff are maintained to handle peak demand periods efficiently.
d) Scenario Planning
The organization prepares for potential disruptions by maintaining buffer capacity in critical operational areas, ensuring resilience and continuity.
e) Digital Tool Integration
Real-time monitoring of projects and resource utilization is enabled through digital platforms, allowing for swift adjustments and improved responsiveness.
f) Communication Prioritization
During execution, regular updates and transparent risk discussions keep internal teams and clients aligned, fostering trust and collaboration.
g) Scalable Buffer Capacity
Manufacturing capabilities are scalable from grams to tons, offering flexibility to meet diverse client needs. This scalability supports the full spectrum of development—from early-phase research to commercial production—without compromising quality or timelines.
By combining data-driven planning, adaptable staffing strategies, and robust communication practices, Neuland consistently meets the needs of multiple biotech clients. This integrated approach ensures that timelines and quality standards are upheld across all engagements.
How do you communicate timeline risks and mitigation strategies effectively to biotech partners?
Neuland is dedicated to building strong, transparent partnerships with biotech clients through the following structured practices:
a) Proactive Risk Communication
Potential timeline risks are communicated early in the project lifecycle, accompanied by clearly defined mitigation strategies. This proactive approach ensures that clients are well-informed and can plan accordingly.
b) Transparent Early Engagement
From the outset, Neuland initiates open discussions around foreseeable challenges—such as supply chain constraints, regulatory shifts, or resource limitations. Where feasible, the potential impact of these challenges is quantified. Clear, jargon-free language is used, supported by visual tools like Gantt charts to outline project milestones, dependencies, and contingency buffers.
c) Structured and Regular Updates
Alignment is maintained through consistent status reports and joint review meetings. These sessions not only highlight emerging risks but also assess the effectiveness of existing mitigation strategies, fostering continuous improvement and agility.
d) Timely Escalation and Resolution Planning
When risks escalate, Neuland’s digitized project management platform enables rapid identification and response. Clients are granted secure access to monitor project progress, view risk statuses, and track resolution plans in real time. This transparency minimizes disruptions and ensures that project momentum, quality, and compliance are maintained.
What practices do you follow to ensure transparency, accountability, and traceability in complex development programs?
Transparency
Neuland fosters transparency by maintaining open and consistent communication with all stakeholders throughout the project lifecycle. This includes sharing updates, challenges, and progress reports using standardized templates and scheduled meetings to ensure everyone remains informed. Documentation is made readily accessible through centralized digital systems, allowing authorized stakeholders to view protocols, batch records, analytical data, and quality reports in real time. Governance structures are clearly defined, outlining roles, responsibilities, and escalation paths to support transparent decision-making and issue resolution. Additionally, risk management is handled proactively, with assessments, mitigation plans, and status updates shared openly to encourage collaborative problem-solving and prevent unexpected setbacks.
Accountability
Accountability at Neuland is reinforced through clearly documented roles and responsibilities, communicated via project charters and RACI matrices. This clarity ensures that individuals and teams understand their ownership of tasks and deliverables. Performance is regularly assessed against agreed timelines and quality standards, with periodic reviews enabling early identification and resolution of concerns. Staff qualifications and competencies are rigorously maintained through detailed training records and assessments, ensuring that all tasks are carried out by skilled personnel. Clients are actively engaged at key decision points and are encouraged to provide ongoing feedback, fostering a culture of mutual accountability and shared success.
Traceability
Neuland ensures comprehensive traceability by implementing robust procedures for documenting, investigating, and resolving deviations, with follow-up on corrective and preventive actions to drive continuous improvement. Accurate and contemporaneous records are maintained across all stages of development—from materials and processes to testing and distribution—aligned with ALCOA+ principles to guarantee data integrity. Any changes to processes, methods, or specifications are managed through formal change control procedures, which include documentation, justification, review, and approval.
This meticulous approach to traceability supports regulatory compliance and reinforces confidence in the integrity of Neuland’s operations.
What’s your approach to method development and tech transfer when receiving incomplete or early-phase packages from biotech’s?
Integrated Development and Scale-Up Approach at Neuland
Neuland initiates its development programs by gaining a deep understanding of reaction mechanisms and kinetics. Through comprehensive process familiarization studies, the team identifies potential gaps and addresses them early, laying the foundation for robust and scalable process design. The analytical team plays a critical role by developing or adapting methods tailored to each stage of development, followed by rigorous validation to ensure precision and reliability.
Technology transfer is streamlined through well-established internal protocols, enabling efficient scale-up from lab to commercial scale. Every step is supported by thorough documentation and detailed reporting, which not only facilitates internal alignment but also strengthens regulatory submissions. This cohesive and methodical approach has consistently enabled early-phase assets to transition smoothly into production, maintaining quality and compliance throughout.
How do you access and communicate your ability to support lifecycle development—from early-phase through to commercialization?
With over forty years of experience in commercial drug substance manufacturing at USFDA-approved facilities, Neuland has established itself as a trusted leader in the development and lifecycle management of active pharmaceutical ingredients (APIs). The company’s extensive track record includes the successful filing of 4 NDAs and 25 IND filings, reflecting its deep regulatory and technical expertise.
Neuland’s robust sourcing capabilities, coupled with a commitment to continuous process optimization, PMI (process mass intensity) reduction and sustainable practices have enabled the implementation of effective process improvement programs. These initiatives have led to the development of lean processes for numerous APIs, enhancing manufacturing efficiency and ensuring long-term commercial viability.
–
Celine Chen1, Dongxin Zhang2
1.Head of European Business, PharmaBlock
2. Senior Director, Tides Center of Excellence, PharmaBlock
At what stage in drug discovery do you recommend CDMO involvement to ensure smoother downstream development?
We recommend involving a CDMO starting from mid to late lead optimization stage, where key decisions around candidate selection (PCC) and initial CMC planning begin to take shape.
During the hit-to-lead stage, biotech teams are primarily focused on rapidly advancing SAR studies. At this point, PharmaBlock often serves as a CRO, delivering high-efficiency compound synthesis. We’ve had clients report that within the same development timeframe, they were able to synthesize 3 times more compounds with PharmaBlock compared to other CROs. This efficiency is largely enabled by our comprehensive building block library, which allows chemists to access a wide variety of pre-qualified intermediates and significantly shorten route design and synthesis timelines.
Once a program enters mid to late lead optimization where potential PCCs start taking shape, it’s critical to bring in a CDMO perspective—even if only for preliminary scalability assessments or route comparisons. In our experience, we’ve seen several biotech programs that had to significantly revise their synthetic routes during Phase II/III. These late-stage changes often result from early decisions that prioritized speed only but overlooked scale-up potential, safety, impurity control, or supply chain feasibility.
At PharmaBlock, our CRO and CDMO teams work in close alignment. We strongly encourage biotech companies to engage CDMO expertise in parallel, beginning with route feasibility evaluations and early CMC strategy. Early CDMO involvement allows us to:
• Assess the synthetic route for scalability, cost sensitivity, and robustness;
• Bridge into impurity profiling, polymorph screening, and analytical method development;
• Build a clear technical transition path from bench chemistry to GMP manufacturing—reducing the risk of rework later on.
Additionally, PharmaBlock offers access to a seasoned CMC advisory team with decades of hands-on experience in global pharma. Our experts have directly led IND and even NDA filings for countless molecules. They support our biotech clients with end-to-end strategic input—across chemistry, analytics, quality, regulatory, and documentation. For many early-stage biotech companies, assembling a full in-house CMC team is not feasible; our team steps in to provide the same caliber of guidance and execution as a large pharma organization.
How do you approach early-stage discussions with biotechs during RFP or feasibility phases?
Working with biotech companies—especially in the early stages—requires a different mindset than supporting large pharma. Most biotechs operate with lean teams, limited data packages, and aggressive timelines. They’re not just outsourcing work; they’re looking for a partner who can bring structure to ambiguity and help them move quickly without compromising quality.
At PharmaBlock, we approach the RFP or feasibility phase as a strategic alignment process. Beyond technical scoping, we invest time in understanding the client’s broader program goals: what drives their next milestone, where timelines are non-negotiable, and how flexible their CMC strategy can be. These early conversations allow us to propose not just “why we can do,” but “what’s most appropriate for the project”.
One topic that often comes up is the use of flow chemistry. While flow offers clear advantages in safety, scalability, and impurity control—especially for challenging or hazardous transformations—we also understand that some biotech clients may be hesitant, especially if unfamiliar with the technology. In such cases, we focus on transparent communication: clearly outlining the expected benefits, potential risks, and timeline implications of using flow versus conventional batch routes. This perspective is built on extensive practical experience—not just theoretical modeling.
In many of our past collaborations, biotech clients who were initially cautious ultimately adopted flow solutions after seeing the data. In fact, we’ve supported multiple cases where a flow-based process developed in early phase was later adopted by big pharma following acquisition, and continued through clinical and commercial supply. For biotech companies seeking funding or exit opportunities, having a robust and scalable CMC package—including well-thought-out process choices—can become a real asset during due diligence.
How do you communicate timeline risks and mitigation strategies effectively to biotech partners?
Once a project is underway, transparency becomes key. We aim to communicate risks early, based on both experimental feedback and institutional experience. For instance, if a synthesis route shows poor scalability or impurity challenges during feasibility, we don’t just flag it—we provide alternative route options, impact estimates, and time recovery plans. For many biotech clients, this kind of proactive risk management is just as valuable as the technical solution itself.
In parallel, we use structured project management tools—regular meetings, risk logs, decision trackers—but equally important is the tone of communication: open, respectful, and technically grounded. We’ve found that building trust early—by asking the right questions, delivering quick wins, and being honest when challenges arise—forms the foundation for effective collaboration throughout the program.
In short, successful biotech partnerships often hinge on clarity at the start and agility along the way. A well-managed feasibility phase sets the tone for the relationship, while transparent risk communication during execution keeps timelines realistic and teams aligned.
How do you support complex modality onboarding (e.g., Peptides, ADCs, PROTACs) from early-phase biotech companies?
Early-phase biotech companies are increasingly driving innovation in complex modalities such as PROTACs, peptides, and ADCs. These molecules often present unique development challenges—such as synthetic complexity, poor solubility, and safety-related manufacturing requirements—that can easily become bottlenecks without the right technical and strategic support.
For many biotechs, it’s not just about finding vendors who can deliver materials, but rather partners who understand how to integrate discovery, CMC, and regulatory considerations early on. For example, in PROTAC programs, the solubility and developability issues commonly associated with these large structures are often rooted in early design decisions. When CRDMOs are brought in early—during lead optimization or even hit-to-lead stages—they can provide insights into synthetic feasibility, physicochemical property optimization, and downstream manufacturability. These early interventions can reduce formulation complexity and accelerate timelines down the road.
At PharmaBlock, we’ve supported a wide range of PROTAC projects, and our experience suggests that integrating discovery chemistry and CMC strategy under one roof can be particularly helpful in this space. Access to one of the largest collections of E3 ligase ligands (both CRBN and VHL), as well as novel linkers of building blocks designed to enhance ADME properties, enables more efficient exploration of SAR and developability trade-offs. Combined with formulation capabilities that address solubility issues—such as amorphous solid dispersions (by spray drying or hot-melt extrusion), nanosuspensions, and lipid-based formulations —this can form a smoother, more informed development path for complex PROTAC molecules.
For peptide therapeutics, we focus on technology innovation to enable the next generation of development. We apply biocatalysis to synthesize unnatural amino acids (UAA), which biotech companies frequently face challenges with. We offer an extensive UAA library, comprising over 10,000 unique compounds, more than 100 of which have been synthesized at scales. Leveraging this vast collection and our know in large-scale UAA production, PharmaBlock empowers peptide biotechs to accelerate their drug discovery timelines and reduce development costs. In parallel, we are advancing new process technologies such as liquid-phase synthesis, and even exploring the use of continuous flow, where PharmaBlock holds recognized technical leadership in the industry. These innovations aim to offer more scalable, cost-effective, reduced PMI for peptide drug manufacturing.
When it comes to ADCs, one of the key challenges for small biotech companies is the lack of access to high-potency containment infrastructure. Collaborating with CDMOs that offer GMP- high-potency capabilities—such as PharmaBlock—can help bridge this gap. We provide safe and reliable synthesis of payloads and linker-payload intermediates, and work seamlessly with biologics partners to support antibody conjugation. This integrated approach enables a more cohesive and efficient development pathway for ADC programs, particularly for resource-constrained biotech teams.
Ultimately, enabling complex modalities isn’t just about solving technical problems—it’s about creating a development strategy that is scalable, cross-functional, and adapted to the pace and resource profile of biotech innovation. CDMOs with integrated capabilities, relevant domain expertise, and the ability to work closely with discovery teams can help de-risk and accelerate these promising programs.
–
Hodaka Shiraishi
Sales & Project Manager, Procos
What is Biotech top three criteria when selecting a CDMO for complex drug modalities like ADCs or oligonucleotides?
When evaluating CDMOs for complex modalities such as ADCs or oligonucleotides, the selection process becomes highly specialized. Usually the top three criteria are:
Technical Competence and Infrastructure:
A proven track record in handling complex chemistry is essential—particularly for high-potency payload synthesis, linker chemistry, and conjugation techniques. The presence of dedicated high-containment suites (OEB-7 (1 ng/m3) compliant), automated conjugation lines, and sophisticated analytical instrumentation.
Regulatory and Quality Compliance:
Biotech looks for CDMOs with a strong history of GMP inspections by authorities such as the FDA, EMA, and PMDA. The Quality Management System (QMS) must be robust, with well-documented SOPs and a culture of continuous improvement. KPI analysis and process enhancement are part of the relationship between the parties, leading to a real partnership. Transparency around audit history and CAPA responses also plays a vital role.
End-to-End Project Integration and Supply Resilience:
The ability to offer a streamlined path from pre-clinical development to commercial manufacturing is critical. CDMOs that can minimize the number of tech transfers and demonstrate resilience in their supply chains (e.g., multiple sourcing strategies) are particularly attractive in fast-to-clinic projects.
Commercial viewpoint:
In the event that Biotech targets commercialization in the foreseeable future, they focus on stable supply and stable company management as well. They check not only technical/quality aspects, but also utilities, financial status, BCP, ESG, CSR, etc.
How vital is CDMO’s track record of on-time project completion, and what evidence do you seek to validate it?
CDMO’s ability to deliver on time is not just important, it’s mission critical. For accelerated development programs, particularly first-in-human or oncology trials, every week counts. Delays can impact clinical enrollment, regulatory submissions, and investor confidence.
To validate delivery performance, Biotech normally requests:
• Project case studies with clearly stated timelines and outcomes.
• Key Performance Indicators (KPIs), typically disclosed during audits.
• References from peer companies.
What key indicators does Biotech check to assess a CDMO’s expertise in handling complex projects such as ADC linker-payload conjugation or oligonucleotide synthesis?
Several tangible indicators help Biotech Companies assess expertise:
• Facility and equipment readiness: Dedicated high-containment areas (e.g., isolators), and advanced synthesis platforms.
• In-house analytical capabilities: The ability to detect residual solvents and impurities, and support stability studies with validated methods is crucial.
• Cross-functional technical teams: The presence of scientific leadership—process chemists, analytical scientists, and formulation experts—with experience in complex modalities is a strong positive signal.
• Governance of the project, KPI & track record, business continuity and project management
How do Biotech determine whether a CDMO has realistic timelines for their project, especially for fast-to-clinic development?
Biotech assesses realism in timelines through multiple lenses:
• Historical data from similar projects, often included in RFI responses or proposals, including proper and clear assumptions to match the requested timelines.
• Critical path analysis shared by the CDMO at the proposal stage, including buffer time for QA, batch release, and tech transfer risks.
• Transparency in resourcing plans, ensuring the project won’t be delayed due to personnel or equipment bottlenecks.
Biotech prefers partners that are honest upfront—even if the timeline is longer—as long as it’s credible.
What red flags indicate a CDMO might struggle with on-time delivery?
Biotech Companies identified several warning signs over the years:
• Frequent scope changes in early discussions, indicating unstable processes or overcommitment.
• Lack of internal manufacturing capacity for key steps.
• Delays in documentation turnaround, such as batch records or change control forms.
• Limited accessibility to critical RMs
• Insufficient disclosure or discussion for technology transfer information
• Frequent personnel changes
Early, direct and open discussions about capacity planning and prioritization policies are useful to mitigate these risks.
During initial due diligence, how do Biotech evaluate a CDMO’s quality management system (QMS)?
Evaluating a CDMO’s Quality Management System (QMS) during due diligence is a foundational step—particularly for projects involving complex modalities. Biotech evaluation typically covers five main pillars:
Inspection History & Regulatory Standing
Biotech begins by reviewing the CDMO’s history of regulatory inspections—FDA, EMA, PMDA—and any Form 483 observations, Warning Letters, or other regulatory actions. A clean inspection track record, or well-documented and timely CAPA implementation, is a strong indicator of QMS maturity.
QMS Structure & Documentation Control
During facility visits or remote assessments, Biotech assesses how the QMS is structured:
• Are there well-defined SOPs covering all cGMP areas?
• Is there a validated electronic Quality Management System in place?
• Is document control centralized, traceable, and audit-ready?
Quality Culture & Governance
Beyond procedures, Biotech assesses the culture of quality. During due diligence, interactions with the QA team provide critical insight into transparency, responsiveness, and mindset.
A risk assessment approach is helpful to keep the QMS reliable and suitable for the customers’ needs.
CDMOs like PROCOS that embrace long-term partnerships and prioritize transparency are more likely to invite customers into their quality governance processes—offering scorecards, quality review meetings, and data sharing. This alignment fosters trust and is a key feature of a “Customer of Choice” model.
–
Fabien Bonhoure
Global Business Project Director, SEQENS
Optimizing drug development: the benefits of early involvement of a CDMO
At what stage in drug discovery do you recommend CDMO involvement to ensure smoother downstream development?
Anticipating the involvement of a CDMO is crucial for ensuring smoother downstream pharmaceutical development. Engaging a CDMO at the transition from drug discovery to pre-clinical development, specifically during the pre-formulation and formulation development stages, provides significant advantages. At this point, CDMOs can help optimize the drug’s chemical and physical properties, anticipate manufacturability challenges, and design robust analytical methods in line with relevant specifications, all of which are essential for later pre-clinical, clinical and commercial speed and success. Early CDMO involvement enables the seamless transfer of technology and knowledge from laboratory to pilot and commercial scale, minimizing potential costly delays and technical setbacks.
For example, when a company developing a novel small molecule involves a CDMO at an early stage, the later can potentially identify different polymorphic profiles that can affect the solubility of the molecule, its formulation and eventually its bioavailability. By optimizing the process to generate a robust and stable polymorph profile, or to master the impurity profile, including genotoxic impurities such as nitrosamine and by developing the related analytical methods upfront, the company can avoid costly reformulation or change in manufacturing process.
Importantly, involving a CDMO during route scouting will allow to select the most robust and cost-efficient process for further scale-up development. Thanks to R&D chemists expertise and capacities, such as solid state characterization, process safety studies, and challenging process parameters (such as critical and operational parameters) by using statistical and modelling tools and high throughput experimentation platform will bring considerable value for time and cost reduction.
At this stage a CDMO can also support in navigating critical regulatory decisions such as the choice of regulatory starting material (RSM) or the definition of the GMP process window.
Choosing the optimal RSM is a complex process that balances regulatory requirements, process control, and manufacturing efficiency. If a CDMO is engaged early, it can apply a science and risk-based framework to justify the selection of starting materials, ensuring that regulatory expectations are met while avoiding unnecessary GMP requirements on upstream intermediates.
For example, if a company selects a RSM too late in the synthetic route for a new drug, regulators can require the company to apply full GMP controls to several upstream intermediates, significantly increasing manufacturing costs and causing a multiple month delay in clinical trial supply. A CDMO with regulatory expertise involved earlier could have advised on selecting an RSM further upstream, balancing regulatory expectations and process efficiency, and avoiding these costly setbacks. CDMO has also knowledge of the RSMs market and therefore can identify the most reliable suppliers for future industrial production.
In summary, the optimal stage for CDMO engagement is immediately after candidate selection and before starting any toxicity & safety studies that can be impacted by the presence of impurities. This proactive approach streamlines the path to pre-clinical, clinical trials and commercialization, ultimately enhancing the probability of success for new drug candidates.
How do you communicate timeline risks and mitigation strategies effectively to biotech partners?
Effective communication of timeline risks and mitigation strategies is fundamental to building trust and achieving success in CDMO-biotech partnerships. The usual communication process begins with a comprehensive risk assessment at project initiation, where all potential technical, operational, and regulatory risks are identified and prioritized based on their likelihood and potential impact. This transparent approach ensures that biotech partners gain insight into potential challenges and understand the rationale behind risk prioritization.
To communicate these risks effectively, it is essential to establish a clear, scheduled communication plan. This includes regular updates, milestone tracking, and review meetings, where progress is measured with predefined KPIs and any emerging risks are openly discussed. Assigning clear responsibilities for each deliverable and risk owner fosters accountability and keeps all stakeholders aligned.
Mitigation strategies should be shared proactively, including contingency plans for high-priority risks, such as alternative technical solutions or backup resources. By continuously monitoring risks and updating partners on both challenges and solutions, the CDMO demonstrates its commitment to transparency and shared problem-solving.
Ultimately, open, structured, and regular communication, anchored by data and clear action plans, enables biotech partners to make informed decisions, respond quickly to challenges, and maintain confidence in the project’s trajectory. Selecting a CDMO that brings end-to-end solutions from route scouting to manufacturing with cutting-edge R&D expertise and capacities is a game-changer.
What’s your approach to analytical method development and tech transfer when receiving incomplete or early-phase packages from biotechs?
When partnering with small or medium-sized biotech or pharma companies, CDMOs often receive incomplete or early-phase technology transfer packages, which can present significant challenges for method development and tech transfer. A comprehensive package ideally includes:
• Detailed chemical process descriptions
• Release specifications for the molecule
• Analytical methods (developed and validated, if available)
• Reference standards
• Impurity profiles or initial assessments
• Safety data sheets (SDS)
• Process flow diagrams
• Batch records
• Stability data
• Regulatory filings or status
• Information on raw materials and critical process parameters
In practice, certain key elements may require further refinement. Common areas for development include ensuring completeness in analytical methods, establishing robust reference standards, thoroughly characterizing impurities and carryover, providing comprehensive safety data, and more detailed process descriptions.
For example, a biotech company might submit a preliminary HPLC method that could benefit from defined system suitability criteria, or a process description that would highlight more details on critical steps and impurity controls. In some cases, chemical processes may currently be optimized for lab-scale application and may need adaptation for safe and efficient scale-up. Additionally, some reagents may pose handling challenges in workshop environments, or initial project assessments may suggest the need for further economic feasibility analysis.
To mitigate the previous risks, our approach begins with a thorough gap assessment: we systematically review the package using a standardized checklist, then organize technical discussions with the client to clarify missing or ambiguous information. If analytical methods are incomplete, our team develops or optimizes them, ensuring phase-appropriate validation. When reference standards are unavailable, we propose sourcing or synthesizing suitable materials. For impurities, we conduct risk assessments and, if needed, design studies to identify, control them and follow their evolution over the process. Up to date ICH guidelines are also applied according to the stage of the development and stability studies are systematically proposed to allow prompt regulatory dossiers filing (IMPD, NDA or CTD).
Transparent communication and collaborative problem-solving are essential. By proactively identifying gaps and working closely with the client, often through regular meetings and shared documentation, we ensure the tech transfer proceeds efficiently, even when starting from an incomplete foundation. This flexible, solutions-oriented approach is critical to meeting aggressive timelines and regulatory expectations in early-phase projects.
–
Jennifer DePolo
General Manager and Site Head of the Siegfried Acceleration Hub
How do you approach early-stage discussions with biotechs during RFP or feasibility phases?
Early discussions are all about alignment and clarity. We start by understanding the biotech’s end goal—whether it’s preparing for an IND, demonstrating early proof-of-concept, or securing funding. From there, we focus on translating that goal into a phase-appropriate development path. One of the key things we emphasize is asking the right technical and strategic questions up front. For example: What are the known characteristics of the molecule? What are the limitations in material supply? What assumptions have already been made about formulation or scalability? These early conversations set the tone for how the program will progress—and help ensure that timelines and investment are well-targeted.
At what stage in drug discovery do you recommend CDMO involvement to ensure smoother downstream development?
Ideally, a CDMO should be engaged before a lead candidate is finalized. That might seem early, but many downstream roadblocks—poor solubility, challenging synthesis routes, or limited scalability—stem from decisions made at the discovery or hit-to-lead stage. Early involvement allows us to provide input on manufacturability and control strategies, while also helping to plan for tech transfer and clinical supply. Even small adjustments at this stage—like choosing more scalable synthetic routes or anticipating GMP needs—can significantly improve the efficiency and robustness of development later.
How do you align your technical team’s input when biotechs bring discovery-stage molecules with limited characterization or formulation data?
In these situations, it’s important to be pragmatic but thorough. We start with a cross-functional assessment involving chemists, formulators, and analytical scientists to identify the data gaps and risk areas. Then, we prioritize experiments that will provide the most decision-driving insight early—solubility screening, forced degradation studies, basic synthetic route assessment, etc.
The aim is to get a minimum viable understanding of the molecule so we can propose a development strategy that is efficient and tailored to phase needs.
If there’s a chance the molecule could be highly potent or a controlled substance, we typically assess it with a degree of conservatism until more is known. As our knowledge of the molecule grows, we adjust controls to be commensurate with the actual risk.
Regular internal alignment ensures that we don’t over-engineer too early but still plan with long-term goals in mind.
How do you support “fast-to-clinic” timelines while maintaining regulatory compliance and phase-appropriate quality?
Speed doesn’t mean skipping steps—it means knowing which steps are essential at which phase. We use a phase-appropriate development framework that helps teams make smart tradeoffs: for example, running smaller parallel experiments or using platform methods where feasible. At the same time, we’re strict about maintaining regulatory compliance. That means building in proper documentation, risk-based decision-making, and early alignment with QA and regulatory colleagues. Having experience across the full development lifecycle helps us guide biotechs on what must be done now—and what can be deferred until it adds real value.
How do you assess and communicate your ability to support lifecycle development—from early-phase through to commercialization?
We begin by evaluating each program’s long-term trajectory at the earliest possible stage, ensuring that development decisions are made with future scalability, tech transfer, and regulatory expectations in mind. This means assessing not only the molecule’s current characteristics but also how the process, formulation, and analytical methods can evolve across the development lifecycle.
To communicate this effectively to our partners, we map out what phase-appropriate development looks like for their specific molecule, outlining how early technical choices—such as synthetic route design or solubility enhancement strategies—can influence manufacturability, stability, and dose flexibility downstream. We also discuss scale-up feasibility, the robustness of analytical methods, and site transfer pathways early on, so expectations are aligned from discovery through to launch.
Ultimately, the goal is to make the early-phase program decisions that avoid rework and keep the long view in sight—reducing risk, cost, and timeline impacts as the molecule progresses.
–
Joachin Fries
Global Head Project Management Drug Products, Siegfried
What should biotechs include in their initial RFPs to better align CDMO capabilities with project expectations?
An effective RFP should clearly outline the scope, including dosage form, batch sizes, timelines, and regulatory goals. But to truly align expectations, it’s just as important to address process-specific needs—such as storage temperatures, sterile handling requirements, and any relevant safety information, including the safety classification of the product.
Biotechs should also indicate whether they’re looking for a partner just for clinical supply or one that can scale to commercial manufacturing. Transferring between providers midstream can introduce delays and risk, so understanding a CDMO’s lifecycle capabilities early can save time later.
It’s helpful to differentiate between “must-haves” and “nice-to-haves”—clarifying which elements are fixed and where there’s room for discussion or process adaptation. And importantly, stay open to CDMO input: experienced partners can offer valuable insights to help design a scalable, commercially viable process from the outset.
Finally, it helps to define expectations for communication, decision-making timelines, and how flexibility should be handled when change inevitably arises.
What key performance indicators (KPIs) should biotechs monitor throughout a CDMO partnership?
Tracking the right KPIs provides visibility and helps keep the project on track. In sterile fill & finish, examples include fill weight accuracy, batch release timelines, deviation rates, and compliance with environmental monitoring thresholds.
Other useful indicators might include lead time for raw material procurement, process deviation resolution time, and success rates on first-time batch releases. For longer projects, schedule adherence and change order turnaround time are also valuable metrics. On-time delivery in full (OTIF)—ensuring products are delivered as ordered, when expected—is a critical supply performance metric, especially as clinical or commercial timelines tighten. Importantly, qualitative factors like responsiveness and documentation quality should also be reviewed regularly—they’re not always reflected in the data, but they often make or break a partnership.
How can biotechs establish more effective communication channels to avoid misalignment with CDMO teams?
Misalignment is most often the result of unclear ownership or inconsistent updates. To avoid this, it’s useful to set up structured governance early—regular joint calls, a shared decision log, and agreed escalation routes. Weekly or bi-weekly meetings, even if brief, can help surface issues before they become delays. Including some in-person meetings can build stronger relationships, improve trust, and foster more effective collaboration.
Using collaborative tools to share documentation and track open items can also support transparency and alignment. During critical phases, increasing the frequency of meetings and setting daily touchpoints can maintain momentum. Ultimately, the key is to foster a working relationship where both sides feel heard and informed—not just during problems, but throughout.
What should biotechs do to prepare their technology transfer packages for smoother collaboration with CDMOs?
Thorough documentation is essential for an efficient tech transfer. This includes data on the formulation, critical process parameters, container-closure systems, and known risks like temperature or light sensitivity. Clearly documenting previous process performance, stability results, and quality attributes helps the CDMO replicate and adapt the process with minimal surprises.
It’s also important to include information on raw material specifications, cleaning validation data (if applicable), and any relevant deviations encountered during development. Finally, designating a single point of contact from both sides to coordinate the transfer—and establishing a joint timeline with review checkpoints—can make a big difference in staying on track.
What advice would you give biotechs about balancing cost, quality, and timelines when selecting a CDMO?
Balancing these three factors is always a challenge. It helps to start by clarifying which trade-offs are acceptable and which are not. For example, speed is often critical for first-in-human studies—but not at the expense of patient safety or regulatory compliance.
Working with a CDMO that has a proven ability to manage sterile manufacturing complexities, such as dosage uniformity and aseptic conditions, can reduce rework and delays down the line. While cost will always be a consideration, underinvesting in quality or skipping early technical due diligence can be more expensive in the long run.
It’s also important to define which elements of your process or timeline are “must-haves” and where there’s room for discussion or flexibility. Being clear about what absolutely must be done versus where you’re open to alternative approaches allows both sides to explore more efficient or cost-effective options without compromising critical objectives.
Ultimately, the right partner should be able to deliver consistent performance across all three dimensions—but alignment on priorities needs to be there from the start.
–
Mat Minardi
EVP of Global Project Management, Sterling Pharma Solutions
What should biotechs include in their initial RFPs to better align CDMO capabilities with project expectations?
The content of RFPs varies greatly, and this can have a very real impact on timelines and the efficiency of a project, alongside the alignment of expectations.
To set a project up for success from the start, the amount of information shared during the proposal process is crucial. This ensures CDMOs can make an accurate and detailed assessment of the project. It is important for biotechs to provide information on the challenges it has faced during the initial stages and what has been tried and failed: this ensures repetitive work is not carried out by the CDMO trying these again.
Gaps in information make a project much harder to judge, and CDMOs then have to factor in additional process and analytical development work into a timeline to try and understand these areas. Any additional steps obviously lead to increased timescales and potentially cost for the biotech.
The other important thing to do during the proposal stage is to set clear direction: for example, are you looking for the CDMO to redesign your process to find efficiencies and look at any challenges there may be as you scale, or do you simply want the CDMO to make your drug substance as fast as possible to the correct specifications?
How can biotechs establish more apparent communication channels to avoid misalignment with CDMO teams?
Clear communication is vital in any partnership, and alignment on what that looks like at the start of the project is important for all parties.
Regular technical meetings which really get into the details of the project are expected – and required – but at Sterling we also initiate smaller steering meetings and check-ins where leaders take the time to step back from the project and look at the bigger picture. This enables transparent communication about project progress, and the customer ensures the goals and objectives remain aligned throughout the project lifecycle.
This may involve the biotech or CDMO bringing in additional disciplines to the conversation, such as regulatory experts, or a biotech’s external consultant to ensure the work being carried out is aligned with the longer-term needs of the project. This forum also serves to discuss what the next steps are, and how the two parties work together to move the project beyond the stage it is currently in.
It is also important to set clear expectations about how you want data to be presented to you, and this does vary on every project. Some customers prefer a presentation to identify key findings, progress and next steps, whereas others want to see every piece of data and report. Most CDMOs would be happy to work either way, but setting these expectations at the start makes everything run more smoothly.
What collaborative approaches have worked for biotechs to build trust and accountability with their CDMOs?
I think the key thing for real partnership working is transparency from all parties involved. We are happy to share everything with the customer including offering an open-door policy to all customers to watch their work being carried out, or something as simple as access to pages from electronic laboratory notebooks (ELNs), if detail is key for its team.
With transparency comes sharing everything during the project; both the highs and the lows. Challenges or issues should be communicated quickly, and should be presented by the CDMO in a way that identifies the issues, outlines impact and then offers potential solutions. The combined team can then work together quickly to find the most appropriate solution.
Flexibility is also part of true collaborative working; development and manufacturing projects rarely stay on a linear journey, so the ability for a CDMO to evolve with a project and be flexible with resources means a project progresses more quickly.
Doing what you say you will do is also something that builds trust within partnerships, if your customer can rely on you, collaboration comes easy. As part of our core value “Be Reliable”, we encourage employees to always be where the customer needs them to be, and to always do the right thing.
–
Rohtash Kumar
Senior Vice President, Chief Technology Officer, Veranova, L.P.
Creating Successful Partnerships for Complex Modalities: Criteria, Timelines, and Tech Transfer Best Practices
Evaluation of CDMOs
What are your top three criteria when selecting a CDMO for complex drug modalities like ADCs or oligonucleotides?
The first criterion is deep knowledge of multiple linker-payload constructs or oligonucleotides from development to commercial manufacturing.
The second criterion is a range of purification, chromatography, and separation technologies that can address the unique challenges of each modality.
The third criterion is comprehensive analytical support for regulatory starting materials, in-process controls, method development, validation, and transfer for intermediates and APIs in compliance with ICH Q2(R1) guidelines.
During initial due diligence, how do you evaluate a CDMO’s quality management system?
A CDMO’s QMS must meet a GMP phase-appropriate approach to support the entire lifecycle of products. The QMS must meet the strictest standards set by regulatory health authorities. In addition, it requires rigorous quality system evaluations, including customer audits and internal corporate and site quality audit programs, which provide continuous and consistent monitoring. The quality system evaluations identify potential areas for improvement and reinforce accountability and transparency within the CDMO. The QMS should ensure data integrity with ALCOA+ principles and validated systems, such as LIMs, MasterControl, and Veeva. Lastly, a track record of continuous improvement focused on increasing efficiency, reducing waste, and improving product quality.
On-Time Completion
How do you determine whether a CDMO has realistic timelines for your project, especially for fast-to-clinic development?
A CDMO has realistic timelines for a biotech project, especially for fast-to-clinic development, if it has a good track record and an effective cross-functional plan to achieve its goals. The CDMO should have a track record of delivering projects on an agreed timeline. The biotech can request from the CDMO recent case studies or similar projects with a detailed timeline. This information should demonstrate the CDMO’s reliability in delivering according to timelines.
Additionally, the CDMO should have experience with fast-to-clinic development and a clear path in place to accelerate the speed to market. This includes a clear statement of work, detailed timelines, and achievable goals. Additionally, a robust chemical development phase is essential for expediting the process and delivery of clinical products while minimizing the need for changes later in the development process. A custom analytical development with a tiered offering supports flexibility and efficiency. Lastly, clear QA standards and in-process control criteria can increase agility and efficiency in production. All these factors play an important role in determining whether a CDMO has realistic timelines for a project.
What red flags indicate a CDMO might struggle with on-time delivery?
A red flag that indicates that a CDMO might struggle with on-time delivery is poor historical performance and lack of clear direction in all necessary functions. Observance of high rates of late delivery, and unrealistic, rushed timelines may cause delays in the overall project timeline and potentially compromise product quality. Furthermore, poor client retention shows dissatisfaction with a CDMO’s services and an inability to meet clients’ expectations, ultimately leading to a lack of repeat business. Poor organization and unclear paths within cross-functional areas including quality and regulatory, project management, production, and technical teams indicate potential struggles may occur in delivery.
Recommendations for Biotechs
What should biotechs do to prepare their technology transfer packages for smoother collaboration with CDMOs?
Biotechs should prepare their technology transfer packages by delivering complete documentation, establishing communication expectations, and assigning an SME as a point of contact for process related questions. The contents of a transfer package may vary depending on the stage of development or manufacturing, but should include a detailed process description, analytical methods, material specifications, batch records, protocols, and relevant regulatory documents.
In addition, there must be a clear communication path established that includes defined points of contact and expectations for regular updates. Lastly, an SME of the process should be available to quickly address questions as the CDMO becomes familiar with the process. These factors can promote a successful technology transfer.
How can biotechs evaluate whether a CDMO’s team has adequate experience and capacity to handle multiple projects?
Biotechs can evaluate if a CDMO has adequate experience by looking at its experience supporting multiple clinical and commercial projects annually. The CDMO should have skilled and appropriately staffed R&D, analytical, quality control, and production teams. These teams are experienced in their respective area and comply with regulatory standards.
The facilities are equally important to the workforce capability in the CDMO’s capacity to handle multiple projects. The laboratories and manufacturing suites must be well equipped with state-of-the-art equipment capable of handling multiple products, and clear capacity planning for efficient use must be in place. The facilities should have sufficient personnel to perform maintenance and minimize equipment downtime. This will allow for multiple projects to be safely and successfully accomplished.

