
Introduction: Background and Significance of PACAP
With the massive success of Semaglutide and related GLP-1 agonists, interest in the therapeutic application of synthetic peptides has skyrocketed.(1-2) Scientists seeking to take advantage of the increased efficacy, selectivity and non-toxicity compared to “small” molecules, have begun to search for more therapeutic peptide feedstocks with refreshed vigor. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) represents one of these promising candidates.(3) The activation of its 3 class B GPCRs, PAC1, VPAC1 and VPAC2 are associated with a myriad of effects such as neuroprotection, cardiovascular regulation, immune responses, metabolic processes, feeding and more.(4, 5) While the therapeutic promise of PACAP is substantial, it suffers from similar drawbacks as most peptide drugs, namely low cell permeability and half-life in vivo.(6, 7) However, recent work as shown glycosylation can negate these issues.(8-13) In fact, PACAP glycopeptide analogues have been shown to more effectively penetrate the Blood-Brain Barrier (BBB) and display effective neuroprotective effects in parkinsonian and ischemic stroke mouse models.(14, 15) This work will cover the design, synthesis, and biophysical properties of the next generation of PACAP glycopeptide analogues.
Design Considerations for Glycosylation. To design effective PACAP analogues, we leveraged the substantial structure-activity relationship (SAR) work available in the literature(16-19) as well as published Cryo-EM data of the native ligand bound to its receptors.(20-22) These previous works demonstrated two key findings: 1) that the first 5-10 N-terminal amino acids are essential for receptor activation and 2) C-terminal attachments can be made with limited effect on efficacy. While previous C-terminal truncations of PACAP showed limited efficacy, this is likely due to poor membrane interactions for which, the addition of the amphipathic behaviors afforded through glycosylation can be addressed.(11) As a result, we have developed PACAP glycopeptide “truncamers”, C-terminal truncations of the PACAP sequence at 10, 14, 19 and 23 amino acids, with an O-linked serine glycoside at the C-terminus. These truncations were made to coincide with one turn of the α-helix to minimize the interaction between the sugar and the extracellular domain of the receptor in its bound conformation. Met17 was also replaced with Norleucine to minimize risk of oxidation and improve shelf life.
Materials and Methods
Synthesis of PACAP Analogues. Synthesis of PACAP “truncamers” analogues followed similar workflow to previously established procedures(14) with several notable exceptions: 1) Deprotection solution was modified to 20% piperidine. 2) Coupling agent was changed to HCTU. 3) All couplings were performed on the Chorus with the DS and DG dipeptides utilizing single shots. 4) Each deprotection step was repeated until a 80% transmittance threshold was achieved.
Membrane Preparation and Protein Identification. Stabled PACAP Receptors (PAC1, VPAC1, VPAC2) at CHO cells were harvested and homogenized for 60 s with a sawtooth homogenizer in 3 mL of membrane buffer (50 mM HEPES pH 7.5). 0.1% of a mammalian protease cocktail inhibitor (Sigma) was added to the buffer immediately before beginning the extraction. Samples were then centrifuged at 4°C for 10 min (1500 rpm) with a Beckman Ultra-centrifuge. The supernatant was isolated and centrifuged again at 4°C for 2 h (32,750 rpm) in Beckman ultra-centrifuge. After centrifugation, the pellet was re-suspended in 1.2 mL of membrane buffer and aliquots of 300 μL were stored at -80°C.(23) Additionally, 50uL were set aside for protein concentration determination. The protein concentration of the membrane was determined using a BCA assay.(24) The purple reaction product was monitored at 560nm using an enzyme-linked immunosorbent assay plate reader (μQuant, Bio-Tek Instruments, Inc.).
Membrane Binding with Cytosolic Protein (G protein, β-Arrestins, etc). The pre-activated (with the compounds) membrane protein is purified and activated on the immobilized surface of the Prisms. And the Prism is settled in the reaction component of PWR (Plasmon Wave-Guided Resonance Spectrometer) (Mainline Scientific, Malvern, PA). The different concentration of the purified Cytosolic Protein is injected into the reaction vessel. The specific shift via the binding of cytosolic protein is recorded. The detail strategy is attached in our earlier publications. The dose response curve will be generated via GraphPad Prism 7. T test is performed to calculate the statistical differences. The data was presented using GraphPad Prism 7.
Synthesis & Analysis of Fmoc Removal
Synthesis of these glycopeptide analogues was accomplished by traditional Solid Phase Peptide Synthesis (SPPS) utilizing a Chorus automated peptide synthesizer. Adaptations compared to previous workflow developed in the Polt group(14) includes a change to HCTU for the coupling agent, 20% piperidine for the deprotection solution and utilization of the UV-Vis Fmoc removal monitoring to continually deprotect until all Fmoc is removed. The SPPS resulted in 12 compounds, each truncation discussed above with a varying degree of glycosylation (unglycosylated, glucoside or lactoside).
While the synthesis is largely traditional (excluding the glycoside), yields are very poor and there is plenty of room for optimization in the PACAP synthesis. To this end, we developed methodology to leverage the UV-Vis data collected during Fmoc removal to better understand the synthetic landscape of these PACAP analogues. The Chorus peptide synthesizer measures transmittance values of the reaction solution as it progresses at 301nm, the local maxima for dibenzofulvene (DBF), the dibenzofulvene–piperidine adduct that is formed by Fmoc cleavage during SPPS. This is often utilized to set a transmission threshold that the reaction solution must obtain before it begins the next coupling, otherwise a subsequent deprotection cycle will occur. However, as difficult deprotections can often translate to difficult couplings(25-27), this collected data represents a facile avenue for information on difficult portions of the synthesis or routes for optimization. To this end, we utilized three analyses of varying involvement to better understand our synthesis.
First, we looked at the average number of deprotection cycles needed for each compound. Analysis of Fmoc removal cycles revealed a glycosylation-dependent increase in synthetic complexity. On average, glucoside analogues required additional deprotection steps, while lactosylated analogues required even more deprotection cycles than the unglycosylated controls for complete deprotection (Table 1).
A positive correlation was also observed between peptide length and deprotection cycles in glycosylated series, particularly for sugar conjugates. This trend was absent in unglycosylated analogues, which consistently required ~3 cycles per residue. These results indicate that both carbohydrate identity and peptide chain length compound the synthetic burden of PACAP analogues.
While average deprotection cycle counts provide a coarse measure of synthetic difficulty, a residue-level analysis is necessary to localize specific bottlenecks during synthesis. To this end, UV-Vis transmittance data that was recorded for each deprotection step was compiled into positional matrices for all PACAP analogues. These datasets capture minimum and maximum transmittance values per residue, enabling comparison across compounds.
Transmittance values were used as a proxy for Fmoc removal efficiency, with the understanding that transmittance is inversely proportional to the concentration of the dibenzofulvene–piperidine adduct in solution. Because each deprotection step will result in a higher transmittance, the transmittance values build on the previous value until the threshold is met. As a result, each transmittance value can inform the effectiveness of the preceding deprotection steps. Therefore, lower transmittance reflects more successful Fmoc removal and potentially more difficult or aggregation-prone regions, while higher transmittance indicates less adduct formation and more accessible cleavage. Since initial deprotection steps universally yielded 0% transmittance, reflecting maximal adduct concentration, only steps 2 and 3 were analyzed in detail (Figure 1).
Heatmap analysis of step 2 revealed that glycosylated analogues consistently exhibited lower transmittance values than unglycosylated counterparts, particularly between residues L21 and Y13, identifying this central segment as an aggregation-prone region. This trend was most pronounced in analogues bearing larger sugar units, suggesting that carbohydrate moieties increase steric and solubility constraints during synthesis. Notably, longer analogues such as TES2320 showed recovery in transmittance values beyond residue 15, implying a possible relaxation of steric hindrance at extended chain lengths.
Step 3 transmittance profiles mirrored those of step 2 but with uniformly higher values, consistent with cumulative Fmoc removal. Importantly, the L21–Y13 low-transmittance region persisted, reinforcing its role as a synthetic hotspot. Unglycosylated analogues again displayed the highest transmittance, supporting the conclusion that glycosylation exacerbates synthetic complexity in a sequence-dependent manner.
While transmittance values can be visualized via heatmaps to assess relative Fmoc removal efficiency, quantitative analysis is also possible through Beer–Lambert’s law, which enables kinetic modeling based on UV-Vis absorbance.
Fmoc removal is generally treated as a first-order reaction, with the rate-limiting step being proton abstraction.(28, 29) The UV-Vis signal corresponds to the concentration of the dibenzofulvene–piperidine adduct, rather than the on-resin Fmoc itself. Given that adduct formation is effectively instantaneous, each deprotection step can be treated as an isolated event, where the maximum adduct concentration inversely reflects the initial Fmoc loading . These data were fitted to exponential decay curves to derive first-order rate constants.
Rate constants were calculated for each second deprotection step across all residues (Figure 2). In cases where the rate constant fell below 0.003 s-¹, the data were more appropriately modeled as 0th-order, suggesting an external rate-limiting factor, likely piperidine diffusion through the resin matrix. Interestingly, residues with more difficult Fmoc removal generally followed first-order kinetics, while more accessible sites displayed zeroth-order behavior.
Analysis of UV-Vis Fmoc removal data has yielded several insights into the synthetic challenges associated with PACAP glycopeptides. Increased glycosylation correlates with greater synthetic difficulty, likely due to hydrophilic aggregation. Specific sequence regions, particularly those exhibiting low transmittance and altered kinetics, were identified as bottlenecks in elongation. Additionally, the variability in Fmoc removal kinetics suggests that reaction order is influenced by local steric or solubility effects.
These findings inform several strategies for optimizing synthesis: (1) enhancing coupling efficiency in problematic regions through stronger reagents, extended reaction times, or multiple couplings; (2) modulating the basicity of the deprotection solution to accelerate cleavage and improve sustainability; and (3) exploring alternative resins to mitigate aggregation-related barriers.
Membrane and Receptor Binding
Understanding the membrane interactions and receptor engagement of PACAP glycopeptide analogues is critical for elucidating their structure–activity relationships. To investigate these dynamics in a physiologically relevant setting, Plasmon Waveguide Resonance (PWR) spectroscopy was employed. This label-free, real-time optical technique is ideally suited for studying membrane-active peptides, offering sensitivity to both mass binding (via p-polarized light) and molecular orientation changes (via s-polarized light) at lipid bilayer interfaces.(30)
PWR operates by coupling polarized light into a planar waveguide composed of a metallic film and dielectric layers, including a supported lipid bilayer or receptor-functionalized membrane mimic (Figure 2). Binding events alter the refractive index and anisotropy at the interface, resulting in measurable polarization-dependent resonance shifts. This dual detection capability enables differentiation between simple surface association and conformational or orientational transitions.(30).
PWR experiments were conducted to characterize glycopeptide interactions with both eggPC lipid bilayers and membranes containing PAC1, VPAC1, or VPAC2 receptors. All analogues demonstrated membrane binding, with p-polarized shifts reflecting mass accumulation and s-polarized changes indicating peptide-induced bilayer reorganization. Notably, glycosylated analogues exhibited reduced membrane affinity relative to unglycosylated counterparts, likely due to increased hydrophilicity conferred by sugar moieties (Table 2).
Further PWR analysis of receptor-embedded bilayers revealed picomolar-range pseudo-EC50 values across all analogues (Table 2). Increased glycosylation correlated with reduced VPAC2 affinity, while PAC1 and VPAC1 binding showed no consistent trend. This may be due to spurious binding of the amphipathic structures to glass and metal surfaces. However, despite this caveat these findings support the conclusion that truncated, glycosylated PACAP analogues maintain high-affinity receptor interactions and can induce conformational changes, providing mechanistic insight into their selective bioactivity.
Outlook for Glycopeptide Drugs
The design and characterization of PACAP glycopeptide analogues presented in this work highlight the promise of glycosylation as a strategy to overcome longstanding challenges in peptide drug development, including limited stability, rapid degradation, and poor bioavailability. Through rational truncation and C-terminal glycosylation, we developed analogues that retain high-affinity receptor engagement while offering improved synthetic tractability and tunable receptor selectivity.
Looking forward, these glycopeptide analogues offer a compelling platform for the next generation of neuroactive therapeutics. Their enhanced solubility and membrane permeability position them well for central nervous system applications, particularly those requiring blood-brain barrier penetration. Moreover, the ability to modulate receptor selectivity, especially through attenuation of VPAC2 activation, opens the door to more targeted therapies with fewer off-target effects.
As glycopeptide synthesis becomes increasingly tractable through data-informed optimization strategies, the broader application of this approach to other peptide hormones and neuromodulators is within reach. Future directions should include in vivo efficacy testing, advanced formulation work (e.g., for intranasal or topical delivery), and further exploration of glycan engineering to fine-tune pharmacodynamics. In an era increasingly defined by biologics, PACAP glycopeptides represent a flexible and powerful scaffold with broad translational potential across neurological, ophthalmic, and metabolic disease landscapes.
Acknowledgements
We thank Gyros Protein Technologies AB for their essential help and access to their PurePep® Chorus
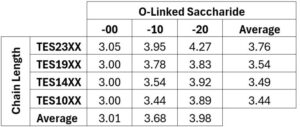
Table 1. Deprotection cycles required for complete Fmoc removal for each PACAP analogue. The first two digits in the compound number indicate the number of PACAP residues in the chain (vertical axis), and the last two digits (horizontal axis) indicate the glycosylation state (00 = ~OH, 10 = β-D-glucoside, 20 = β-lactoside).
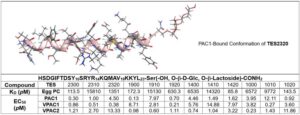
Table 2. Binding Assay of PACAP Glycopeptides towards EggPC and membrane with highly expressed PAC1, VPAC1,VPAC2 receptors. KD, (N=6) is the dissociation constant, is the concentration of peptides required for half of its binding sites on a membrane or receptors to be occupied at equilibrium. (Details and binding curves are provided in the Supplemental Information.)

Figure 1. Heatmaps of step 2 minimum (A) and maximum transmittance values (B)compared to step 3 minimum (C) and maximum transmittance values (D) of each amino acid for PACAP analogues.

Figure 2. Calculated rate constants for the 2nd deprotection step of all amino acids.
References and notes
- Peptide Therapeutics Market: Global Industry Analysis, Size, Share, Growth, Trends and Forecast 2012−2018. Albany. NY: Transparency Market Research; 2012.
- Wang L, Wang N, Zhang W, Cheng X, Yan Z, Shao G, et al. Therapeutic peptides: current applications and future directions. Signal Transduction and Targeted Therapy. 2022;7(1):48.
- Cherait A, Xifró X, Reglodi D, Vaudry D. More Than Three Decades After Discovery of the Neuroprotective Effect of PACAP, What Is Still Preventing Its Clinical Use? J Mol Neurosci. 2025 Jun 21;75(3):80. doi: 10.1007/s12031-025-02366-z. PMID: 40542938; PMCID: PMC12182478.
- Holland PR, Barloese M, Fahrenkrug J. PACAP in hypothalamic regulation of sleep and circadian rhythm: importance for headache. The Journal of Headache and Pain. 2018;19(1):20.
- Gargiulo AT, Curtis GR, Barson JR. Pleiotropic pituitary adenylate cyclase-activating polypeptide (PACAP): Novel insights into the role of PACAP in eating and drug intake. Brain Research. 2020;1729:146626.
- Lau JL, Dunn MK. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic & Medicinal Chemistry. 2018;26(10):2700-7.
- Diao L, Meibohm B. Pharmacokinetics and Pharmacokinetic–Pharmacodynamic Correlations of Therapeutic Peptides. Clinical Pharmacokinetics. 2013;52(10):855-68.
- Mabrouk OS, Falk T, Sherman SJ, Kennedy RT, Polt R. CNS penetration of the opioid glycopeptide MMP-2200: a microdialysis study. Neurosci Lett. 2012;531(2):99-103.
- Hay M, Polt R, Heien ML, Vanderah TW, Largent-Milnes TM, Rodgers K, et al. A Novel Angiotensin-(1-7) Glycosylated Mas Receptor Agonist for Treating Vascular Cognitive Impairment and Inflammation-Related Memory Dysfunction. J Pharmacol Exp Ther. 2019;369(1):9-25.
- Werle M, Bernkop-Schnürch A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids. 2006;30(4):351-67.
- Apostol CR, Hay M, Polt R. Glycopeptide drugs: A pharmacological dimension between “Small Molecules” and “Biologics”. Peptides. 2020;131:170369.
- Bernard K, Mota JA, Wene P, Corenblum MJ, Saez JL, Bartlett MJ, et al. The angiotensin (1–7) glycopeptide PNA5 improves cognition in a chronic progressive mouse model of Parkinson’s disease through modulation of neuroinflammation. Experimental Neurology. 2024;381:114926.
- Christina Hoyer-Kimura. PNA5, A Novel Mas Receptor Agonist, Improves Neurovascular and Blood-Brain-Barrier Function in a Mouse Model of Vascular Cognitive Impairment and Dementia. Aging and disease. 2024;15(4):1927-51.
- Apostol CR, Bernard K, Tanguturi P, Molnar G, Bartlett MJ, Szabo L, et al. Design and Synthesis of Brain Penetrant Glycopeptide Analogues of PACAP With Neuroprotective Potential for Traumatic Brain Injury and Parkinsonism. Front Drug Discov (Lausanne). 2022;1.
- Bernard K, Dickson D, Anglin BL, Leandro Heien M, Polt R, Morrison HW, et al. PACAP glycosides promote cell outgrowth in vitro and reduce infarct size after stroke in a preclinical model. Neurosci Lett. 2024;836:137883.
- Bourgault S, Vaudry D, Botia B, Couvineau A, Laburthe M, Vaudry H, et al. Novel stable PACAP analogs with potent activity towards the PAC1 receptor. Peptides. 2008;29(6):919-32.
- Bourgault S, Vaudry D, Segalas-Milazzo I, Guilhaudis L, Couvineau A, Laburthe M, et al. Molecular and conformational determinants of pituitary adenylate cyclase-activating polypeptide (PACAP) for activation of the PAC1 receptor. J Med Chem. 2009;52(10):3308-16.
- Doan ND, Letourneau M, Vaudry D, Doucet N, Folch B, Vaudry H, et al. Design and characterization of novel cell-penetrating peptides from pituitary adenylate cyclase-activating polypeptide. J Control Release. 2012;163(2):256-65.
- Apostol CR, Tanguturi P, Szabo LZ, Varela D, Gilmartin T, Streicher JM, et al. Synthesis and In Vitro Characterization of Glycopeptide Drug Candidates Related to PACAP(1-23). Molecules. 2021;26(16).
- Wang J, Song X, Zhang D, Chen X, Li X, Sun Y, et al. Cryo-EM structures of PAC1 receptor reveal ligand binding mechanism. Cell Research. 2020;30(5):436-45.
- Duan J, Shen D-d, Zhou XE, Bi P, Liu Q-f, Tan Y-x, et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nature Communications. 2020;11(1):4121.
- Liang Y-L, Belousoff MJ, Zhao P, Koole C, Fletcher MM, Truong TT, et al. Toward a Structural Understanding of Class B GPCR Peptide Binding and Activation. Molecular Cell. 2020;77(3):656-68.e5.
- Lipkowski AW, Kosson P, Misicka A, Bonney I, Toth G, Hruby VJ, et al., editors. Endomorphin as a template for development of new opioids agonist-tachikinin antagonist chimeric ligands2006: JOHN WILEY & SONS LTD THE ATRIUM, SOUTHERN GATE, CHICHESTER PO19 8SQ, W ….
- Fillebeen C, Descamps L, Dehouck M-P, Fenart L, Benaı̈ssa M, Spik G, et al. Receptor-mediated Transcytosis of Lactoferrin through the Blood-Brain Barrier *. Journal of Biological Chemistry. 1999;274(11):7011-7.
- Larsen BD, Holm A. Incomplete Fmoc deprotection in solid-phase synthesis of peptides. Int J Pept Protein Res. 1994;43(1):1-9.
- Behrendt R, White P, Offer J. Advances in Fmoc solid-phase peptide synthesis. J Pept Sci. 2016;22(1):4-27.
- Collins JM, Singh SK, White TA, Cesta DJ, Simpson CL, Tubb LJ, et al. Total wash elimination for solid phase peptide synthesis. Nature Communications. 2023;14(1):8168.
- Ralhan K, KrishnaKumar VG, Gupta S. Piperazine and DBU: a safer alternative for rapid and efficient Fmoc deprotection in solid phase peptide synthesis. RSC Advances. 2015;5(126):104417-25.
- Luna OF, Gomez J, Cárdenas C, Albericio F, Marshall SH, Guzmán F. Deprotection Reagents in Fmoc Solid Phase Peptide Synthesis: Moving Away from Piperidine? Molecules (Internet). 2016; 21(11).
- Rascol E, Villette S, Harté E, Alves ID. Plasmon Waveguide Resonance: Principles, Applications and Historical Perspectives on Instrument Development. Molecules (Internet). 2021; 26(21).

