
49
Introduction
Peptides are an important kind of biomolecule with an increasing importance in drug discovery, (1) cosmetics, (2) agriculture, (3) and material science, (4) among others. This privileged position of peptides in the scientific world, with a strong impact on the markets, is undoubtedly thanks to the development of solid-phase synthesis (SPS) led by Nobel Prize winner R. Bruce Merrifield in the 1960s (5). Without the strategy of solid-phase peptide synthesis (SPPS), peptides would be solely the subject of academic study with some applications as biochemical tools.
In SPPS, the peptide elongation takes place with the growing peptide attached to a solid support/resin. This allows a very easy work-up because the excess reagents and soluble side-products can be removed by simple washing and filtrations. Using solid-phase chemistry, the SPPS process is highly streamlined; the method is an iterative cycle of adding reagents dissolved in the appropriate solvents, followed by filtration and adding solvents for washing until a final cleavage and elution step. This allows automation, which has sped up much of all the peptide research.
For many years, SPPS has been carried out at room temperature (6). However, it is important to take into account that in SPPS, the reagents (amino acids, coupling and deprotecting agents) need to diffuse into the polymeric resin beads to reach the reactive sites (7). Higher temperatures reduce solvent viscosity and increase the rate of diffusion, ensuring that the reagents can effectively reach all available reaction sites within the resin matrix. In addition, SPPS performed at higher temperatures enables the reduction of deprotection and coupling times by speeding up reaction kinetics (8), (9). Heat reduces peptide aggregation on the resin, which causes most of the side reactions; reducing aggregation optimizes the accessibility of free amino acids of the growing peptide chain for subsequent addition steps (10).
Meeting the rising demand for peptide therapeutics and for other applications requires the development of methodologies that increase the productivity of commercial-scale peptide manufacturing facilities. At small-medium scale, several approaches have demonstrated success at improving synthesis speed, including microwave heating, (11) rapid stirring, (12) and variable bed flow reactors; (13) these methods have not been demonstrated to meet the demands of large commercial scale production.
Conduction heating is a low-cost, simple to integrate method of providing heat to peptide synthesis processes. It provides precise, uniform temperature control, in contrast to other heating methods that may potentially generate hotspots (11). As synthesis quality is sensitive to temperature, maintaining tight control over the temperature of the reagents matters for optimizing the full process, reducing the need for double coupling by improving the reaction kinetics and minimizing undesirable reactions such as racemization. Conduction heating can be implemented at all scales, streamlining process adaptation as demands on throughput increase.
Here, we integrated a heat exchanger on the CSBio 136X, a research scale automated peptide synthesizer, to allow preheating of reagents prior to transfer to the heated reaction vessel. Preheating was expected to reduce the time required for reagents to reach the target temperature, speeding up synthesis time, while enabling deprotection and coupling reactions to occur at higher temperatures to improve product yield and purity.
Materials and Methods
Peptides were synthesized on a CSBio 136X peptide synthesizer modified to include a heat exchanger. Peptides were synthesized at a 0.1 mmol scale on Rink amide polystyrene resin (loading 0.67mmol/g) in a 20 mL jacketed reaction vessel using DIC/Oxyma as the coupling reagent. Fmoc amino acids were prepared to a final concentration of 0.3M in 0.3M Oxyma in dimethylformamide (DMF). Diisopropylcarbodiimide (DIC) was prepared to a final concentration of 0.3M in DMF. Fmoc deprotection was achieved with 20% piperidine in DMF.
The performance of the heat exchanger was established by setting the temperature of the circulator bath and measuring the temperature of the solvent upon reaching the reaction vessel. The heat exchanger was integrated such that the delivery of any solvents, reagents, or solutions to the heated reaction vessel would be first pre-heated. The heated reaction vessel was jacketed and provided uniform heating throughout the synthesis with the same circulator that was heating the heat exchanger.
Initially, all peptides were synthesized at room temperature with 60 min of coupling and two successive deprotection steps, 5 and 10 min, for comparison with the heated synthesis. Jung-Redemann (JR) peptide synthesis was performed at 60 °C and 80 °C, with 60 min of coupling and 5 + 10 min of deprotection; subsequent syntheses were performed at 80 °C with 10 min of coupling and 5 and 10 min deprotection steps. ABRF-1992 peptide synthesis was performed at 60 °C with 10 min of coupling and 2 + 5 min deprotection. The Bradykinin peptide was synthesized at 60 °C with 10 min of coupling and 1 + 3 min of deprotection. Oxytocin was synthesized under two conditions: 40 °C and 60 °C with 10 min coupling and 1 + 3 min deprotection times. Peptides were cleaved from the resin using a standard TFA/TIS/H2O solution (95:2.5:2.5) for 1.5 h. The unprotected peptides were precipitated with cold ether and the solid dissolved in 10% aqueous acetic acid (AcOH) and lyophilized.
Reduction of the Met sulfoxide on JR peptide was performed prior to cleavage. The peptidyl-resin was treated with a mixture of 20 eq. of Me3SiCl and 2 eq. Ph3P (3 × 1 h) in THF (14). After cleavage, the Met alkylation was reverted by keeping the crude peptide in 5% acetic acid at 40 °C for 24 hours (15).
Cleaved peptides were analyzed by LCMS in an UltiMate 3000 UHPLC-ISQ EC single quadrupole mass spectrometer in positive-ion mode (Thermo Fischer Scientific Inc). Chromatography was performed on a Phenomenex AerisTM C18 column (3 µm, 4.6 × 150 mm), with a flow rate of 1.0 mL/min. All peptides were eluted in a gradient over 15 minutes. Buffer A consisted of 0.1% Formic Acid (FA) in H2O and Buffer B was 0.1% FA in acetonitrile (CH3CN). The JR peptide was eluted over a gradient from 15% to 60% Buffer B; the ABRF 1992 peptide was eluted over a gradient from 5% to 40% Buffer B; the Bradykinin peptide was eluted over a gradient from 0% to 40% Buffer B; and finally, Oxytocin was eluted over a gradient from 10% to 40% Buffer B. Data were analyzed using ChromeleonTM (CDS) Software (version 7.3).
Results and Discussion
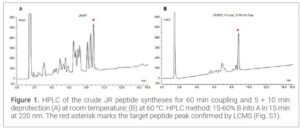
JR peptide (H-WFTTLISTIM-NH2) was selected as a model. Even being a sequence consisting just of 10 amino acids, its synthesis is challenging due to its tendency to aggregate. The first synthesis was attempted at RT with 1h coupling and 5+10 min for Fmoc removal. The results (Figure 1A) align with expectations, as it is well known that this peptide, although short in size, aggregates and tends to produce low-quality crude peptides; in this case, the purity was 13% (16). A new JR peptide synthesis was performed by increasing the temperature to 60 °C. For this purpose, all reagents were preheated through a heat exchanger prior to delivery to the jacketed reaction vessel of the CSBio 136X that was kept at the same temperature for 60 min of the coupling reaction and 5 + 10 min for Fmoc removal. Under these conditions, the purity of the crude increased considerably, reaching a 39% purity (Figure 1B).
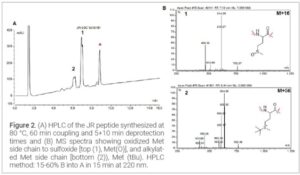
Further increasing the synthesis temperature to 80 °C apparently resulted in worsening the quality of the final peptide (Figure 2A). However, the main impurity of a mass of +16 (Figure 2A, peaks 1 and Figure 2B top), corresponds to the conversion of Met sulfide to sulfoxide (Met(O)). The second impurity (Figure 2A, peaks 2 and Figure 2B bottom) is due to the tert-butylation of the Met (Met(tBu)). Both impurities appear as a double peak because of the chirality of the sulfoxide and sulfonium moieties. We hypothesize that the sulfoxidation might be due to the high temperature (80 °C) and the long coupling time, however the t-butylation might be unrelated to the high temperature because this side reaction is associated with the cleavage reaction.
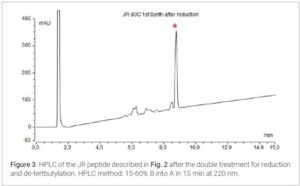
These two side reactions can be reversed. The sulfoxide of Met can be reduced to the disulfide before the final global deprotection and cleavage by treatment with Me3SiCl and Ph3P,(14) and the tert-butylation can be removed after cleavage by treatment with 5% AcOH at 40 °C for 24 hours.(15) Thus, the HPLC (Figure 3) after this double treatment renders a crude peptide of better purity than that obtained at 60 °C.
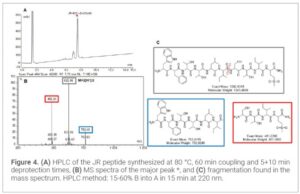
The JR peptide synthesis was repeated under the same conditions, 60 min coupling and 5+10 min deprotection at 80 °C to check the reproducibility of the oxidation pattern. In this case, the major product found showed a mass of M+32, which indicates a double oxidation. The JR peptide sequence not only contains the Met residue susceptible to oxidation but also could occur on the Trp residue. However, the unusual fragmentation of the peptide showed us that the +32 mass was due to the double oxidation of Met to form the corresponding sulfone that cannot be reverted to the desired product (Figure 4).
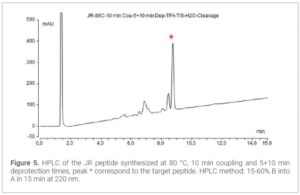
We decided to repeat the JR synthesis, keeping the temperature at 80 °C and the same deprotection time but with a reduced coupling time. The purpose of this experiment was to determine if the large extent of oxidation on Met was taking place during the long coupling or it was favored during the deprotection. Thus, the coupling time was reduced from 60 min to 10 min. The analysis of JR peptide crude by LC-MS showed a main peak which had the mass of the desired peptide (Figure 5). None of the other peaks had a mass corresponding to single or double oxidation on Met, rather, the mass found for them corresponded to deletions of several amino acids. This result confirms that the oxidation took place mainly due to the long coupling reaction at a high temperature of 80 °C. The crude purity in this case was 48%, which represents a great improvement compared to the 13% achieved at RT. Moreover, increasing the temperature from RT to 80 °C, decreased the total synthesis time from over 13 hours down to 5 hours.
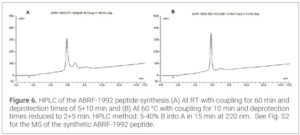
To continue the exploration of the impact of increasing the synthesis temperature on the final purity of crude peptide, we synthesized other model peptides. First, we synthesized the 16 amino acid peptide proposed by the Association of Biomolecular Resource Facilities (ABRF) in 1992 (17) to evaluate peptide synthesis methods among its members (ABRF-1992, H-GVRGDKGNPGWPGAPY-NH2). Two syntheses of ABRF-1992 peptide were carried out, the first one under standard conditions, RT with 60 min for coupling and 5 + 10 min for Fmoc removal, and the second at 60 °C with just 10 min for coupling and 2 + 5 min for the deprotection. Figure 6 shows that the second synthesis, at higher temperature and reduced reaction times, resulted in a peptide with clearly higher purity. Importantly, the time for the overall synthesis was reduced from 22 to 5.6 hours.
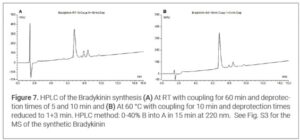
The next peptide we studied was Bradykinin (H-RPPGFSPFR-NH2). We chose this short peptide to evaluate if the high temperature had any influence in the deletion of Arg due to δ-lactam formation. The first synthesis was run under our standard conditions (RT with 60 min for coupling and 5 + 10 min for Fmoc removal) for comparison with the new conditions, synthesis carried out at 60 °C, 10 min coupling and 1 + 3 min for Fmoc removal. When analyzed, the crude peptide obtained after each synthesis showed two main impurities in the HPLC traces whose mass corresponded in both cases to the deletion of Arg (Figure 7). However, contrary to expectations, the integration of those peaks was less in the case of the synthesis at 60 °C with shorter coupling and deprotection times. The overall purity of the crude improved from 70 % under standard conditions to 84% with the new ones. Additionally, the time of the full synthetic process was reduced from 12 h to 3 h.
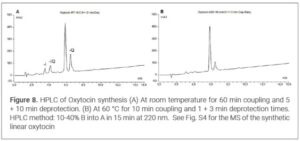
Finally, we performed the synthesis of Oxytocin (H-CYIQNCPLG-NH2) as a model to study the effect of increasing temperature during the full process on Cys residues. The synthesis performed under standard conditions generated several deletion peptides (Ile, Gln, and of both, Ile-Gln). In the next synthesis, the temperature was increased moderately to 40 °C and times were reduced to 10 min for coupling and 1 + 3 min for deprotection. The improvement in the quality of the crude was not significant, but increasing the temperature to 60 °C, using the same time as before, nearly eliminated the generation of deletion peptides. Furthermore, the total synthetic time was reduced from 12 hours to less than three hours using the solvent preheating system (Figure 8).
Conclusion
This work demonstrates that conduction heating-assisted SPPS is an excellent technique for preheating solvents and reagents prior to delivery to the jacketed reaction vessel, enabling faster, more efficient coupling and deprotection steps and therefore increasing the quality of the crude peptide and decreasing the time required to perform peptide synthesis. Shortly, these conditions improved the quality of the crude peptide in less time.
However, the use of higher temperatures and longer reaction times can result in some side-reactions, therefore, it is recommended that reaction times be reduced when using high temperatures.
With the industry’s focus towards reducing PMI (process mass index) and shifting away from DMF, maintaining strict temperature control will be essential for efficient solvent use in SPPS. Temperature directly affects solvent viscosity, which in turn dictates the time required for every step of the process. Moreover, the kinetics of coupling and deprotection are influenced by temperature and can differ considerably depending on the solvent used. Thus, conduction heating-assisted SPPS can facilitate the use of other solvents.
This strategy of solvent preheating is expected to be useful at research scale, and, importantly, can also be readily scaled up to meet the demands of commercial-scale synthesis. Using this methodology of precise control over temperature may enable us to adjust the use of traditional solvents on our synthesizers and explore the effect of temperature control on the use of alternative green solvents.
As a final remark, the heat exchanger allows for the close monitoring and management of synthesis temperature in ways that other heating technologies cannot. The obtention of higher crude purities enables the simplification of downstream purification and overall reduces the necessity for additional syntheses and materials, and more importantly, purifications to generate the desired mass of product.
References and Notes
This work was originally presented as a poster at IOPC 2025 in Prague, Czech Republic.
Mason, S., Kumar, A., Carpiz, B., Greiner, W., de la Torre, B. G., Albericio, F., Slavazza, D., & Chang, H. (2025). A Conduction Pre-Heating System to Optimize Solid-Phase Peptide Synthesis (Poster presentation). IOPC 2025
- Muttenthaler M, King GF, Adams DJ, Alewood PF. Trends in peptide drug discovery. Nature reviews Drug discovery. 2021;20(4):309-25. https://www.nature.com/articles/s41573-020-00135-8
- Pintea A, Manea A, Pintea C, Vlad R-A, Bîrsan M, Antonoaea P, et al. Peptides: Emerging Candidates for the Prevention and Treatment of Skin Senescence: A Review. Biomolecules (Internet). 2025; 15(1). https://www.mdpi.com/2218-273X/15/1/88
- Zhang Y-M, Ye D-X, Liu Y, Zhang X-Y, Zhou Y-L, Zhang L, et al. Peptides, new tools for plant protection in eco-agriculture. Advanced Agrochem. 2023;2(1):58-78. https://www.sciencedirect.com/science/article/pii/S2773237123000035
- Das TN, Ramesh A, Ghosh A, Moyra S, Maji TK, Ghosh G. Peptide-based nanomaterials and their diverse applications. Nanoscale Horizons. 2025;10(2):279-313. https://pubs.rsc.org/en/content/articlelanding/2025/nh/d4nh00371c
- Merrifield RB. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. Journal of the American Chemical Society. 1963;85(14):2149-54. https://pubs.acs.org/doi/10.1021/ja00897a025
- Ferrazzano L, Catani M, Cavazzini A, Martelli G, Corbisiero D, Cantelmi P, et al. Sustainability in peptide chemistry: current synthesis and purification technologies and future challenges. Green Chemistry. 2022;24(3):975-1020. https://pubs.rsc.org/en/content/articlehtml/2022/gc/d1gc04387k
- Martin V, Jadhav S, Egelund PHG, Liffert R, Johansson Castro H, Krüger T, et al. Harnessing polarity and viscosity to identify green binary solvent mixtures as viable alternatives to DMF in solid-phase peptide synthesis. Green Chemistry. 2021;23(9):3295-311. https://pubs.rsc.org/en/content/articlelanding/2021/gc/d1gc00603g
- Pacini L, Muthyala M, Aguiar L, Zitterbart R, Rovero P, Papini AM. Optimization of peptide synthesis time and sustainability using novel eco-friendly binary solvent systems with induction heating on an automated peptide synthesizer. Journal of Peptide Science. 2024;30(9):e3605. https://onlinelibrary.wiley.com/doi/full/10.1002/psc.3605
- Grunhaus D, Molina ER, Cohen R, Stein T, Friedler A, Hurevich M. Accelerated Multiphosphorylated Peptide Synthesis. Organic Process Research & Development. 2022;26(8):2492-7. https://pubs.acs.org/doi/10.1021/acs.oprd.2c00164
- Tamás B, Willi PL, Bürgisser H, Hartrampf N. A robust data analytical method to investigate sequence dependence in flow-based peptide synthesis. Reaction Chemistry & Engineering. 2024;9(4):825-32. https://pubs.rsc.org/en/content/articlelanding/2024/re/d3re00494e
- Pedersen SL, Tofteng AP, Malik L, Jensen KJ. Microwave heating in solid-phase peptide synthesis. Chemical Society Reviews. 2012;41(5):1826-44. https://pubs.rsc.org/en/content/articlelanding/2012/cs/c1cs15214a
- Alshanski I, Bentolila M, Gitlin-Domagalska A, Zamir D, Zorsky S, Joubran S, et al. Enhancing the Efficiency of the Solid Phase Peptide Synthesis (SPPS) Process by High Shear Mixing. Organic Process Research & Development. 2018;22(9):1318-22. https://doi.org/10.1021/acs.oprd.8b00225
- Sletten ET, Nuño M, Guthrie D, Seeberger PH. Real-time monitoring of solid-phase peptide synthesis using a variable bed flow reactor. Chemical Communications. 2019;55(97):14598-601. https://pubs.rsc.org/en/content/articlehtml/2019/cc/c9cc08421e
- Nandhini KP, Albericio F, de la Torre BG. 2-Methoxy-4-methylsulfinylbenzyl Alcohol as a Safety-Catch Linker for the Fmoc/tBu Solid-Phase Peptide Synthesis Strategy. The Journal of Organic Chemistry. 2022;87(15):9433-42. https://pubs.acs.org/doi/10.1021/acs.joc.2c01057
- Nandhini KP, Alhassan M, Veale CGL, Albericio F, de la Torre BG. Methionine-Containing Peptides: Avoiding Secondary Reactions in the Final Global Deprotection. ACS Omega. 2023;8(17):15631-7. https://pubs.acs.org/doi/10.1021/acsomega.3c01058
- Carpino LA, Krause E, Sferdean CD, Schümann M, Fabian H, Bienert M, et al. Synthesis of ‘difficult’ peptide sequences: application of a depsipeptide technique to the Jung–Redemann 10- and 26-mers and the amyloid peptide Aβ(1–42). Tetrahedron Letters. 2004;45(40):7519-23. https://www.sciencedirect.com/science/article/abs/pii/S0040403904017010?via%3Dihub
- Fields GB, Carr SA, Marshak DR, Smith AJ, Stults JT, Williams LC, et al. Evaluation of Peptide Synthesis As Practiced in 53 Different Laboratories. In: Angeletti RH, editor. Techniques in Protein Chemistry IV: Academic Press; 1993. p. 229-38. https://www.sciencedirect.com/science/article/abs/pii/B9780120587575500305?via%3Dihub

