
Introduction
Peptide therapeutics have emerged as a critical class of pharmaceuticals, with over 60 peptide drugs approved globally and a market projected to reach USD 57 billion by 2027. Projections (1) suggest the GLP-1 market will reach $100-150 billion by 2030 and 9% of the U.S. population will be using GLP-1RAs by 2030. The success of GLP-1 receptor agonists has fueled demand for large-scale peptide production, necessitating innovations in synthetic technologies. Traditional synthetic methods, particularly SPPS, face challenges related to high solvent consumption, poor atom economy, and environmental concerns. Recent efforts have focused upon greener, more efficient, and scalable approaches to meet regulatory and market demands. This manuscript highlights aspects of SPPS, LPPS, tag-assisted, hybrid, and continuous manufacturing of peptides. (2)
SPPS
SPPS was introduced by Merrifield in the 1960s and synthesis of peptides is accomplished by attaching amino acids step-by-step to grow the peptide chain which is tethered to an insoluble solid support (resin). Each cycle involves deprotection (removing a protecting group from the amino acid) and coupling (adding the next amino acid). (3) Once the peptide sequence is completed, it is cleaved from the resin and purified. This technique revolutionized peptide chemistry and the obvious advantages of SPPS include automation: SPPS is highly compatible with automated synthesizers, which handle repetitive deprotection and coupling steps with precision; efficiency and speed: SPPS is fast because it uses excess reagents to drive reaction to completion reducing overall process times compared to liquid-phase peptide synthesis (LPPS). Complex peptides can be synthesized in days rather than weeks; simplified purification: the solid support acts like a handle, keeping the peptide anchored while reagents and by-products are washed away. This minimizes the need for intermediate purification until the final step, typically high-performance liquid chromatography (HPLC); flexibility: SPPS allows the incorporation of non-natural amino acids, modifications (e.g., pegylation, fatty acids, etc.), and enables cyclic peptides. The well recognized disadvantages of SPPS include: high solvent consumption (e.g., DMF, NMP, dichloromethane) which raises environmental concerns; batch-to-batch variability and processes that limit scalability for commercial production; purification steps that are resource-intensive and expensive; high manufacturing risk: one mistake during an SPPS process can cause entire batch failure. Recent advances in SPPS, particularly from 2020 to 2025, have focused on coupling and deprotection strategies that can provide greater sustainability through automation, higher efficiency, and expansion of synthetic capabilities to meet the growing demand for complex peptides.
Improvements in Fmoc/tBu Chemistry
The 9-fluorenylmethoxycarbonyl (Fmoc)/tert-butyl (tBu) strategy remains the dominant approach in SPPS due to its milder conditions compared to the Boc/benzyl strategy. However, impurities can arise during synthesis, purification, and storage that impact the safety, efficacy, and quality of final product. Effective management of impurities involves identification, characterization and control strategies in adherence with regulatory guidelines. Some recent advancements have been achieved regarding side reaction mitigation strategies (4) and various in-silico tools can now predict difficult peptide sequences based upon their physicochemical properties and sequence motifs. These tools help anticipate common problems like aggregation, incomplete couplings and impurity formation, allowing for optimized synthetic protocols. Analyzing the peptide sequence beforehand to predict synthetic challenges can identify problematic regions (e.g., sterically hindered or aggregation-prone regions) and adjustments can be made to the synthetic strategy.
Side reaction mitigation techniques serve to prevent truncation or deletion (5) in peptide synthesis, where the peptide chain terminates prematurely or has an omission in the desired sequence. Optimizing coupling using higher-quality reagents ensures amino acids, coupling reagents (e.g., HBTU, HATU, or DIC/HOBt), and solvents (e.g., DMF, NMP) are pure and free of contaminants and excess reagents can drive reactions to full completion. Double coupling for difficult residues (e.g., arginine, histidine, or β-branched amino acids like isoleucine) or performing triple coupling cycles can ensure complete incorporation. Pre-activation of amino acids reduces side reactions and use of tests (Kaiser, ninhydrin, TNBS or chloranil) can confirm complete couplings after each cycle and microcleavage of the peptide from a portion of the resin for full HPLC analysis can provide detailed intermediate insight.
Choice of appropriate protection groups (e.g., Fmoc or Boc) is important for stability and to avoid side reactions like aspartimide formation. Maintaining optimal temperatures (typically 20–30°C for Fmoc SPPS) can also prevent side reactions. Sequence-dependent issues like hydrophobic or β-sheet-forming sequences that lead to aggregation can be prevented by pseudo-proline dipeptide use, backbone-protecting groups (e.g., Hmb or Dmb), or low-substitution resins. When handling difficult amino acids like cysteine, methionine, or tryptophan (prone to oxidation or alkylation), appropriate protective groups or inert atmospheres can prevent side reactions. (6) Implementing capping protocols on unreacted N-terminal amines with acetic anhydride to block further chain elongation on incomplete sites can prevent deletion impurities from propagating. This also aids purification by altering the retention times of any capped deletions.
Optimizing deprotection and cleavage conditions using mild deprotection reagents avoid side reactions while ensuring deprotection and adding scavengers to TFA cocktails can trap reactive species and prevent alkylation or other modifications that could mimic deletions.
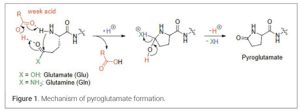
Pyroglutamate Formation (7)
Pyroglutamate (also known as pyroglutamic acid or pGlu) formation is a major side reaction in peptide synthesis where N-terminal glutamine (Gln) or glutamic acid (Glu) cyclizes. This involves the N-terminal amine attacking the side-chain carbonyl, leading to deamination and a five-membered lactam ring. It can occur during deprotection, cleavage, or storage, reducing yield and purity. (Figure 1)
If possible, avoid placing Gln or Glu at the N-terminus as this position is most susceptible to cyclization. Instead, incorporate these residues internally or use conservative substitutions (e.g., replace Gln with Asn or another polar amino acid) to maintain functionality while reducing risk. Acetylating the N-terminal Gln (e.g., Ac-Gln-…) can block the free amine group preventing cyclization under acidic cleavage conditions and can be done post-synthesis or during SPPS if necessary. Employing bulky protecting groups for the side chain of Gln or Glu, can sterically hinder cyclization while also assisting in suppressing pyroglutamate formation.
Aspartimide formation (8), Diketopiperazine (DKP) formation (9), Guanidinylation (10), and Racemization (11) can also be minimized via similar optimizations in protecting group strategy, additives, or through reduced exposure to piperidine, optimized coupling and cleavage conditions, and sequence specific strategies.
Advances in Green and Sustainable SPPS
Green chemistry and process efficiency is a growing focus, addressing the future sustainability of peptide synthesis. Environmental impact of hazardous solvents like DMF and NMP, toxic coupling by-products, safety and performance of reagents, and reactive process additives all provide opportunities for greener and safer peptide processing. Advances in aqueous peptide synthesis use water-soluble protecting groups like 2,7-disulfo-9-fluorenylmethoxycarbonyl (Smoc), can achieve yields of 61–71% for di- to decapeptides. (12) Coupling reagents like COMU with 2,6-lutidine or TCFH with collidine have demonstrated improved efficiency under aqueous conditions. (13).
Solvent Reduction
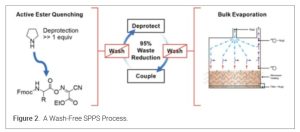
A 2023 breakthrough report on Nature Communication claimed that solvent-intensive washing steps could be eliminated by using volatile Fmoc deprotection bases and bulk evaporation with headspace gas flushing, reducing waste by up to 95% while maintaining peptide quality. (14) (Figure 2).
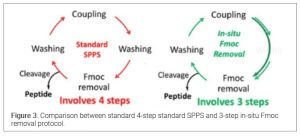
The traditional coupling cycle for SPPS always follows 4 steps: 1) coupling reaction with DIC/OxymaPure, 2) washes (5-6 times) with DMF to remove excess of reagents and side products; 3) Fmoc removal with bases (20% PIP in DMF); 4) washes (5-6 times) with DMF to remove excess PIP and dibenzofulvene and its adduct. The copious washing represented 80-90% of the total waste from SPPS processing. It may be possible to eliminate washes if upon coupling reaction completion, 20% PIP in DMF is added directly (in-situ) for Fmoc removal without additional washes. After Fmoc removal is completed, 2-3 resin washes can be added with 1% OxymaPure in DMF to remove the excess PIP, DBF and its adduct. This protocol (15) takes advantage of the mild acidity of the OxymaPure to remove all basic piperidine and as a result, the number of DMF washing steps are greatly reduced to minimize the overall solvent consumption (16) (Figure 3).
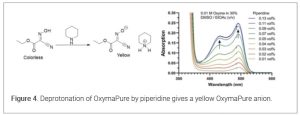
Piperidine removal after Fmoc deprotection is important as residual piperidine leads to double insertion of amino acids due to unintentional Fmoc-removal of the incoming amino acid derivative, or to deletions related to sequestering of the activated amino acid derivative which forms the corresponding piperidine amide. A PAT method using UV-vis for monitoring residual piperidine during the DMF washes after an Fmoc removal step found OxymaPure was deprotonated by the base and formed a yellow solution at an absorption band in the 400−550 nm region. This is not surprising as K-Oxyma displayed an absorption band of similar shape to that of OxymaPure itself in the presence of piperidine (in the 400−550 nm region), supporting the idea that the OxymaPure anion is the chromophore. (16) (Figure 4)
This is a real-time online UV−vis monitoring system to quantify piperidine with high sensitivity. In addition to potential reduced washing, this tool allows determination of completed washing by visual inspection for yellow color of OxymaPure in the presence of piperidine.
Greener solvents including binary solvents (17) are being considered as replacements for DMF but challenges persist such as: resin swelling ability (4-7 mg/g), solubility of amino acid building blocks and coupling reagents (>0.25M), melting point and solvent viscosity < 4 mPa*s, coupling kinetics (< 1h), Fmoc removal and epimerization. Studies have identified greener alternatives, but there is not yet a single solvent that fulfills all requirements. Binary solvent systems can be tailored to accomplish these requirements. (18) A tool (19) to identify viable binary solvent mixtures via a polarity-viscosity plot has been presented and includes ten binary solvents mixtures in the “green SPPS zone” all containing DMSO with different co-solvent ratios. Coupling reactions proved superior using low polarity, binary solvent systems while Fmoc removal performed better with higher polarity binary solvents.
There are also alternative bases for Fmoc removal (20), and reports of superior coupling reagents, (21-24) including phosphonium-based reagents such as BOP, PyBrOP, PyAOP, PyOxim, and PyBOP which offer rapid coupling (often complete in minutes) with minimal racemization. These reagents react with a base to form highly reactive species that generate active esters (e.g., OBt or OAt esters) for efficient coupling. As the demand for peptide-based drugs grows, coupling reagents will remain at the forefront of SPPS advancements, driving precision and sustainability. N- to C-peptide synthesis has also emerged as a sustainable alternative to conventional C- to N-SPPS, minimizing protecting groups and excess reagents. (25) Techniques for precipitation, filtration and drying without lyophilization can also provide improvements. (26)
LPPS
LPPS is preferred for shorter peptides (≤10–15 amino acids) as it offers advantages in scalability and cost for certain sequences. LPPS avoids solid support, minimizing waste from resin washing and disposal. Recent advancements in LPPS include the use of greener solvents and improved coupling reagents to enhance reaction efficiency such as fast, widely applicable and green solution-phase peptide synthesis (GSolPPS) via a continuous protocol using propylphosphonic anhydride as the coupling reagent and N-benzyloxycarbonyl-protecting group (Z), which is easily removed by hydrogenation. Lower PMI of ~30 is possible by reducing intermediate purification, which has been applied to hybrid strategies for GLP-1 drugs (27).
Hybrid Peptide Synthesis Development (GLP-1 Peptide)
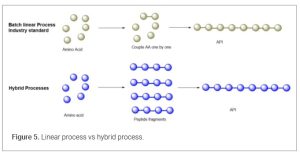
The rapid development and massive demand for GLP-1 receptor agonists (like semaglutide, tirzepatide, and others) have significantly accelerated advancements in peptide synthesis technologies. GLP-1 drugs require gram-scale to kilogram-scale production of complex peptides (20–40+ amino acids). SPPS linear synthetic protocols alone are no longer an industry standard for manufacturing peptide drug substance for pre-clinical and clinal studies. SPPS protocols, because of multiple sequential operations, often lead to additional risk related to potential manufacturing failure. If something fails during the linear sequence the entire batch may be compromised. Industry reports suggest as high as a 25% failure rate for linear approach peptide synthesis. (28) Hybrid synthesis combines SPPS and LPPS to overcome their individual limitations. In this approach, SPPS is used to synthesize peptide fragments, which are then coupled in solution via LPPS or ligation techniques. The synthesis of Fuzeon (29) (enfuvirtide), a 36-amino-acid peptide, on metric ton scale has demonstrated the viability of hybrid methods. For GLP-1 drugs, hybrid synthesis enables the production of long, complex sequences with high purity, addressing SPPS’s length constraints and LPPS’s solubility challenges. Recently, Eli Lilly’s manufacturing facility in Cork, Ireland, employed a hybrid SPPS-LPPS platform to produce Tirzepatide, achieving a production capacity of metric tons per year. (30) In this hybrid approach, SPPS is used to synthesize peptide fragments (6–14 amino acids), which are then coupled via LPPS. This reduces production times and enables parallel synthesis at multiple global supply nodes, enhancing supply chain resilience. (Figure 5)
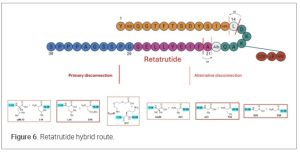
LPPS is effective for continuous processing as well, such as using plug flow reactors, which can replace traditional batch methods. These reactors enable continuous peptide coupling, reducing equipment footprint and capital costs while improving scalability. (31) Hybrid peptide approaches take advantage of SPPS to achieve high purity on shorter fragment peptide chain and synthesis of peptide fragments in parallel decreasing manufacturing risk while offering a more convergent approach. It also provides improved analytical control as fragments of the peptide can be monitored for purity prior to commitment to the final peptide production. Fragment condensation in a liquid phase setting can provide higher yield while reducing solvent consumption. A bonus is that the final condensation of fragments can often be performed in traditional small molecule equipment and facilities. It is critical to couple the fragments at non-epimerizable amino acid residues to prevent racemization. The fragment isolated from SPPS must have good physical properties including precipitation during the fragment isolation and acceptable solubility in fragment condensation solvents. This hybrid process reduces process cycle time and improves overall efficiency. A recent talk (32) from Eli Lilly gave a glimpse of future peptide manufacturing. Retatrutide is a 39-mer peptide and first in class GIP, GLP-1, and Glucagon receptor tri-agonist. The fragment strategy is critical for the overall process. The key element of fragment strategy is coupling with a non-epimerizable amino acid residue at C-terminus. The team from Lilly investigated different fragment strategies to ensure all fragments are well behaved after soft cleavage. The fragments isolated from soft cleavage are all white powders with good solubility in the condensation solvent systems used. For the fragment condensation in liquid phase, the team investigated 10 options (Figure 6). The study also found that Aib loaded CTC-resin was shown to be highly labile with OxymaPure used for coupling reaction.
Tag-Assisted Peptide Synthesis
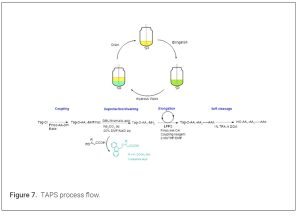
Tag-assisted peptide synthesis (TAPS) enhances SPPS or LPPS by incorporating chemical tags to improve solubility, purification, or coupling efficiency. These tags, often hydrophobic or hydrophilic groups, are temporarily attached to the peptide chain and removed after synthesis. (33) Early examples include the use of hydrophobic trialkoxyl-benzyl alcohols for synthesizing growth hormone-releasing peptides, combining LPPS with disulfide bond formation. The development began around 2005 for peptide synthesis, with formal reports in 2012 detailing a fluorene-derived anchor for efficient liquid phase peptide synthesis by using Fmoc/Boc protocol as with SPPS. (34) TAPS offers cost-effectiveness, scalability, and environmental benefits. It reduces reagent consumption by 5-8 times versus SPPS, using near-stoichiometric reagents and greener solvents like CPME or EtOAc. Solvent use can drop by 60%, with PMI (process mass intensity) often lower than SPPS. Homogeneous reactions improve coupling efficiency for sterically hindered amino acids, yielding >90% purity without chromatography. Peptide CDMOs have been actively involved in the development of tag assisted peptide synthesis (TAPS), and there were several talks in 2025 Tides USA related to this technology. Tag-assisted peptide synthesis has transformed LPPS into a viable, sustainable alternative to SPPS, enabling efficient production of therapeutic peptides. With technologies like AJIPHASE® leading commercial adoption, TAPS is poised to meet the growing demand for peptides in medicine, emphasizing green chemistry principles. Despite advantages, challenges include tag design for very long or hydrophobic peptides, where solubility issues persist. Cleavage conditions may cause side reactions, and some tags (e.g., PEG) complicate analysis due to polydispersity. Scalability for long peptides (>15 residues) remains limited. Future developments may integrate automation and flow chemistry for continuous TAPS to further reduce waste. Greener tags from renewable sources and hybrid SPPS-TAPS methods could address limitations. (Figure 7)
Continuous Manufacturing (35,36)
High PMI and production cycle times lead to significantly higher cost of complex APIs. LPPS technology has the potential to significantly reduce costs, cycle time, and PMI with favorable reaction kinetics suitable for use in a general-purpose API manufacturing plant. In batch SPPS, reactions occur in discrete vessels with manual or semi-automated steps for coupling, deprotection, and washing, leading to extended cycle times and waste generation from excess reagents required to drive reactions to completion. Continuous flow provides a streamlined process that enables in-line monitoring, precise control of temperature and pressure, reduced residence times, and reduced reagent use. Flow systems can be solid-phase (CF-SPPS), (37,38) where the peptide remains anchored to a solid support in a packed-bed reactor, or liquid-phase, where synthesis occurs in solution with soluble support or tag-assisted peptide synthesis. CF-SPPS adapts traditional SPPS by passing reagents through a resin-packed column reactor. Key innovations include automated fast-flow peptide synthesis (AFPS), which uses heated activation loops and variable-bed reactors to monitor resin swelling and deprotection efficiency via UV-Vis spectroscopy. In AFPS, couplings can occur at rates of 25 per hour, enabling synthesis of peptides up to 164 amino acids. Another approach involves mesoscale reactors operating at elevated temperatures (e.g., 70°C) and pressures (60 bar), reducing coupling times to 6-7 minutes and Fmoc-deprotection to 1.5 minutes. Recent developments incorporate microwave-assisted heating or deep learning models trained on UV-trace data from thousands of deprotection steps to predict aggregation and optimize sequences in real-time. (39) Continuous flow offers improved efficiency and speed with amino acid incorporation occurring every 1.8 minutes under automated control, drastically reducing synthesis time. In-line spectroscopic monitoring ensures precise control and efficient mixing. (40) Flow methods require only 1.5 eq. of amino acid for quantitative conversions, compared to 4-10 eq. for batch, lowering costs and waste. Liquid-phase flow further minimizes solvents by enabling greener alternatives like water-compatible activators. (41) Flow aligns with green chemistry principles by reducing chemical waste and energy use, while addressing the high environmental impact of traditional SPPS (e.g., DMF solvent usage) (42).
There are several challenges in continuous flow peptide manufacturing (43) such as clogging and heterogeneity, pressure drops in reactors necessitating sonication or variable-bed designs, purification and washing efficiency, catalyst and device limitations, deactivation of immobilized catalysts, and regulatory and adoption barriers. Continuous flow represents a paradigm shift in peptide manufacturing, but scalability and compliance currently remain limited.
Macrocyclic Peptide and Stapled Peptide Synthesis and Manufacturing
Macrocyclic peptides, (44) characterized by their often large, cyclic molecular structures, have emerged as a promising class of therapeutics due to their high potency, selectivity, and potential for oral bioavailability compared to traditional linear peptides. These molecules, typically comprising 8–40 amino acid residues with one or more cyclic domains, offer a unique balance of small-molecule drug-like properties and biologics’ specificity. Stapled peptides (45) are conformationally constrained α-helical peptides with hydrocarbon “staples” that enhance stability, cell permeability, and binding affinity. Macrocyclic peptides broadly encompass any cyclic peptide, including head-to-tail cyclized, sidechain–to–sidechain linked, and hybrid cyclized systems. Both approaches seek to stabilize bioactive conformations while improving proteolytic stability and pharmacokinetics. Stapled and macrocyclic peptides have emerged as powerful scaffolds for drug discovery, especially for targeting protein–protein interactions (PPIs) that are often “undruggable” by small molecules. However, their complex structures pose significant challenges for synthesis and large-scale manufacturing. Recent advancements, such as enlicitide decanoate (MK-0616), an oral PCSK9 inhibitor, demonstrate transformative approaches to overcoming these challenges. (46)
Impact upon CDMO Resourcing
The exponential growth of manufacturing required for GLP-1 peptides has revolutionized the peptide industry and ushered in a time of industrial focus upon creative, efficient, and cost-effective processes that satisfy larger production. This has resulted in a renewed interest and application of combined synthetic and isolation strategies that can deliver quality peptides in a sustainable and industrially feasible manner. These efforts have had a very positive impact upon the industry, but programs outside of the GLP-1 franchises have experienced some negative impacts from these changes.
Traditional peptide manufacturers have realized the dispersion of experienced peptide scientists who have chosen to explore lucrative opportunities in support of GLP-1 programs. This has resulted in a shallowing of the relative experience at CDMO’s regarding peptide development and manufacture. Fortunately, the void is being filled by new scientists with renewed interest in pursuing peptide synthesis related to the expanding career opportunities illustrated by GLP-1 volumes and challenges.
Another impact has been the ability of GLP-1 programs to preferentially obtain capacity at manufacturers. This does impact CDMO resource and capacity availability for other peptide programs. In addition, some critical product activities (like fill/finish) have experienced large industrial suppliers being purchased by GLP-1 innovators to ensure dedicated capacity. Fortunately, many suppliers have completed or are in process of expanding capacity and personnel to meet the needs of the industry as a whole and some GLP-1 innovators are building large plants of their own to ensure supply chain robustness which may reduce capacity pressure on CDMO’s.
Peptides represent great promise as therapeutics and successes suggest an expanding need for suppliers to support pharmaceutical industry innovators. It is not yet certain that the CDMO industry has achieved the ability to deliver against this great opportunity.
Challenges and Future Directions
The success of GLP-1 drugs has catalyzed a renaissance in peptide synthetic technologies to address the need for sustainable, scalable, and efficient production. Innovations in SPPS, including PMI reduction and greener solvents, alongside LPPS, tag-assisted synthesis, hybrid approaches, peptide engineering, and automation/digitization, have transformed the field. These advancements not only support the growing demand for peptide therapeutics but also align with global sustainability goals. Despite advancements, challenges remain in peptide synthesis. SPPS’s high PMI and reliance on hazardous solvents require continued innovation in greener alternatives and recycling technologies. LPPS’s limitations for longer peptides necessitate further development of hybrid and tag-assisted methods. Automation and digitization, while promising, require significant investment, limiting adoption in smaller facilities. Regulatory pressures, such as stricter impurity limits, demand improved analytical technologies. Future directions include continuous flow SPPS, fully aqueous synthesis, and integration of artificial intelligence to streamline manufacturing processes. Continued research and industrial collaboration will further enhance the efficiency and environmental footprint of peptide manufacturing, ensuring the continued expansion of peptides as pharmaceuticals.
References and notes
- Morgan Stanley Research (2025). The Exponential Growth of Obesity Drugs; McKinsey & Company (2025). What are GLP-1 medications?
- Martin , P. H. G. Egelund , H. Johansson, Sebastian T.L. Quement , F. Wojcik and D. S. Pedersen* RSC Adv., 2020, 10, 42457-42492
- Amblard M, Fehrentz J, Martinez J, and Subra G. (2006). Methods and Protocols of Modern Solid Phase Peptide Synthesis. Molecular Biotechnology, 33(3), 239–254
- Behrendt R, Whiteb P, Offerc J. Advances in Fmoc solid-phase peptide synthesis, J Pept Sci. 2016 Jan 20;22(1):4–27
- Guzman, F. “Introduction to Peptide Synthesis.” Curent Protoc Protein Science. (2001). AAPPTec. “Practical Synthesis Guide to Solid Phase Peptide Chemistry.” (PDF, Accessed 2025)
- Amblard, M., Fehrentz, J. A., Martinez, J., & Subra, G. (2006). “Methods and protocols of modern solid phase peptide synthesis.” Molecular Biotechnology, 33(3), 239–254
- Coin, I., Beyermann, M., Bienert, M. (2007). “Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences.” Nature Protocols, 2(12), 3247–3256. Bierne G, Weller M. Investigation of Impurities in Peptide Pools. Separations 2025, 12
- Kong M, Braak T, Neumann K. Aspartimide Formation and Its Prevention in Fmoc Chemistry Solid Phase Peptide Synthesis. ChemBioChem 2025
- Yang Y, Hansen L. Optimized Fmoc-Removal Strategy to Suppress the Traceless and Conventional Diketopiperazine Formation in Solid-Phase Peptide Synthesis, ACS Omega 2022, 7, 12015−12020.
- Vrettos E, Sayyad N, Mavrogiannaki E, Stylos E, Kostagianni A, Papas S, Mavromoustakos T, Theodorou V, Tzakos A. Unveiling and tackling guanidinium peptide coupling reagent side reactions towards the development of peptide-drug conjugates. RSC Adv., 2017, 7, 50519–50526
- Duengo S, Muhajir M, Hidayat A, Musa W, Maharaniet R. Epimerization in Peptide Synthesis. Molecules 2023, 28, 8017
- Knauer S, Koch N, Uth C, Meusinger R, Avrutina O, and Kolmar H. Sustainable Peptide Synthesis Enabled by a Transient Protecting Group, Angew. Chem. Int. Ed., 2020, 59, 12984–12990
- Jaradat D, Musaimi O, and Albericio F. Advances in solid-phase peptide synthesis in aqueous media (ASPPS) Green Chem., 2022, 24, 6360–6372
- Collins J, Singh S, White T, Cesta D, Simpson C, Tubb L, Houser C. Total wash elimination for solid phase peptide synthesis, Nature Communications | (2023) 14:8168
- Mthethwa N, Nandhinia N, Kumar A, Sharma A, Torre B and Albericio F. Toward sustainable solid-phase peptide synthesis strategy-in situ Fmoc removal. Green Chemistry Letters and Reviews, 2024, 17, 1, 2325993
- Broman S, Rosenberg M, Wojcik F, Hansen A, Egelund P, Malmstro̷m J, Pedersen D. Green Solid-Phase Peptide Synthesis: Oxyma-Triggered Spectrophotometric Monitoring of Residual Piperidine, Org. Process Res. Dev. 2024, 28, 666−673
- Ferrazzano L, Catani M, Cavazzini A, Martelli G, Corbisiero D, Cantelmi P, Antoni T, Mattellone A, Luca C, Felletti S, Cabiri W., Tolomelli A. Sustainability in peptide chemistry: current synthesis and purification technologies and future challenges. Green Chemistry, 24, 975–1020, 2022
- Martin V, Egelund P, Johansson H, Quement S, Wojcik F and Pedersen S. Greening the synthesis perspective of peptide therapeutics: an industry perspective, RSC Adv., 2020, 10, 42457
- Jadhav S, Castro H, Martin V, Egelund P, Krüger T, Richner F, Albericio F, Pedersen F. Replacing DMF in solid-phase peptide synthesis: varying the composition of green binary solvent mixtures as a tool to mitigate common side-reactions, Green Chem., 2021, 23, 3312
- Martelli G, Cantelmi P, Palladino C, Mattellone A, Corbisiero D, Fantoni T, Tolomelli, A. Replacing piperidine in solid phase peptide synthesis: effective Fmoc removal by alternative bases, Green Chem., 2021, 23, 8096–8107
- F, Ei-Faham A. Choosing the Right Coupling Reagent for Peptides: A Twenty-Five-Year Journey. Org. Process Res. Dev. 2018, 22, 760−772
- McFarland A, Buser J, Embry M, Held C, and Kolis S. Generation of Hydrogen Cyanide from the Reaction of Oxyma/DIC, Org. Process Res. Dev., 2019, 23, 2099
- Ferrazzano L, Catani M, Cavazzini A, Martelli G, Corbisiero D, Cantelmi P, Antoni T, Mattellone A, Luca C, Felletti S, Cabri W., Tolomelli A. Sustainability in peptide chemistry: current synthesis and purification technologies and future challenges. Green Chemistry, 24, 975–1020, 2022
- Nzama H, Manne S, Marder O, Orosz G, Torre B, Albericio F. Unveiling the quaternary carbodiimide symphony: harmonizing green chemistry in peptide synthesis, GREEN CHEMISTRY LETTERS AND REVIEWS 2024, VOL. 17, NO. 1, 239282
- Ghosh K, Lubell W. N- to C-Peptide Synthesis, Arguably the Future for Sustainable Production, Journal of Peptide Science, 2025; 31: e70019
- Kopach M, Development of a Three Fragment Hybrid SPPS/LPPS Synthesis for Retatrutide, Boulder Peptide Symposium, 2024
- Mattellone A, Martelli G, Corbisiero D, Ferrazzano L, Cantelmi P, Palladino C, Tolomelli A and Cabri W. Speeding Up Sustainable Solution-Phase Peptide Synthesis Using T3P® as a Green Coupling Reagent: Methods and Challenges. Green Chemistry, 25, 2563–2571, 2023
- Isidro-Llobet A, Kenworthy, M, Mukherjee S, Kopach M, Wegner K, Gallou F, Smith A, Roschangar F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615−4628
- Schneider, S.; Bray, B.; Mader, C.; Friedrich, P.; Anderson, M.; Taylor, T. Development of HIV fusion inhibitors, J. Peptide Sci. 11: 744–753 (2005)
- Anya H. Disruptive Innovations Shaping the GLP-1 Landscape, PDA Letter, Jul 16, 2025
- Schneider, S; Bray, B.; Mader, C; Friedrich, P.; Anderson, M; Taylor, T. et al. Kilogram-Scale GMP Manufacture of Tirzepatide Using a Hybrid SPPS/LPPS Approach with Continuous Manufacturing, Org. Process Res. Dev. 2021, 25, 1628−1636
- Michael Kopach, Development of a Three Fragment Hybrid SPPS/LPPS Synthesis for Retatrutide, Boulder Peptide Symposium, 2024
- Sharma A, Kumar A, Torre B, Albericio F. Liquid-Phase Peptide Synthesis (LPPS): A Third Wave for the Preparation of Peptides. Chem. Rev. 2022, 13516-13546
- Novel diphenylmethyl-Derived Amide Protecting Group for Efficient Liquid-Phase Peptide Synthesis: AJIPHASE, Org. Lett., Vol. 14, No. 17, 2012
- Simona M, Heiderb P, Zoua Y, Liaoa X, Alexander M, Pentelute B. et al. Rapid Flow-Based Peptide Synthesis, Chembiochem. 2014 March 21; 15(5): 713–720
- Otake Y, Adachi K, Yamashita Y, Iwanaga N, Sunakawa H, Shamoto T, Itoh H. et al. A liquid-phase continuous-flow peptide synthesizer for preparing C-terminal free peptides, React. Chem. Eng.,2023, 8,863
- Kubo D, Fuse Y, Otake Y, Adachi K, Hattori Y, Ogawa J. Yokogawa Technical Report English Edition V 11 .66 No.1 (2023)
- Ruhl, K, Di Maso, M, Rose, H, Schultz, D, Lévesque, F, Grosser S, Silverman, S, Li, S, Sciammetta, N, Mansoor, U. Continuous-Flow Solid-Phase Peptide Synthesis to Enable Rapid, Multigram Deliveries of Peptides. Org. Process Res. Dev. 2024, 28, 2896−2905
- Nina Hartrampf et al. Chimia 75 (2021) 480–483
- Hartrampf N., Saebi A., Poskus M., Gates Z., Pentenlute B. Synthesis of proteins by automated flow chemistry, Science, Vol 368, issue 6494, 980-987, 2020
- Mándity I, Olasz B, Ötvös S, Fülöp F. et al. Continuous-Flow Solid-Phase Peptide Synthesis: A Revolutionary Reduction of the Amino Acid Excess. Chemsuschem, 2014, vol 7, issue 11, 3172-3176
- Somehsaraie M, Vavsari V, Kamangar M, Balalaie S. Chemical Wastes in the Peptide Synthesis Process and Ways to Reduce Them. Iran J Pharm Res. 2022 December; 21(1): e123879
- Schiefelbein K, Hartrampf N. Flow-based Methods in Chemical Peptide and Protein Synthesis. CHIMIA 2021, 75, No. 6, 480
- Gang D, Kim D, Park H. Cyclic Peptides: Promising Scaffolds for Biopharmaceuticals. Rev. 2023, 123, 4971–5042
- Schafmeister, C. E.; Po, J.; Verdine, G. L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. Am. Chem. Soc. 2000, 122, 5891–5892
- Li, H.; Thaisrivongs, D. A.; Shang, G.; et al. Total Synthesis of Enlicitide Decanoate. J. Am. Chem. Soc. 2025, Online Early Access

