
COMMENTARY ARTICLE
Redefining Therapeutics through Targeted Protein Degradation
Beyond inhibition: the transformative power of targeted protein degradation
Targeted Protein Degradation (TPD) represents a paradigm shift in drug discovery by leveraging cellular proteostasis mechanisms to remove disease-relevant proteins instead of merely inhibiting them. Unlike traditional small-molecule inhibitors, TPD strategies—such as PROTACs and molecular glues—enable the catalytic degradation of previously undruggable targets. These modalities are rapidly advancing through preclinical and clinical pipelines, with over 20 degraders currently in trials and applications that span oncology, immunology, neurodegeneration, and rare diseases, along with increasing uses. Strategic partnerships and venture investments, totaling billions of dollars, underscore confidence in TPD’s scalability and commercial viability. This panel discussion explores the evolution of TPD from a scientific curiosity to a transformative therapeutic engine, addressing challenges such as resistance, tissue specificity, and synthetic complexity. We highlight recent innovations, including stimuli-responsive degraders and cell-specific delivery systems like Antibody-PROTAC conjugates, as well as AI-driven design. TPD is no longer a conceptual promise—it is reshaping modern pharmacology by transforming protein degradation into a programmable and precision-guided intervention strategy.
The Degrader Revolution: Moving Beyond Inhibition
Conventional small-molecule inhibitors function by occupying an active site to block enzymatic function or modulate protein activity. However, 80–85% of the human proteome lacks suitable binding pockets, rendering most disease-relevant proteins pharmacologically inaccessible through traditional methods. TPD provides a powerful alternative.
Proteolysis-targeting chimeras (PROTACs) and molecular glues represent two leading TPD strategies. These molecules combine a protein of interest (POI) and an E3 ubiquitin ligase, facilitating ubiquitination and subsequent degradation by the 26S proteasome. This event-driven mechanism contrasts with occupancy-driven inhibition, allowing for catalytic activity—once the target is degraded, the degrader is released to repeat the cycle.
The implications are far-reaching: PROTACs and related TPD technologies can degrade previously intractable targets such as transcription factors, scaffolding proteins, and protein–protein interaction hubs. Additionally, the catalytic nature of degradation enables lower dosing, prolonged pharmacodynamic effects, and the potential to overcome resistance that plagues many small-molecule inhibitors.
Recent innovations—including stimuli-responsive PROTAC prodrugs, nanoparticle delivery systems, and expansion beyond CRBN and VHL to novel E3 ligases—improve selectivity, bioavailability, and safety. These next-generation degraders are not just incremental upgrades; they represent a reengineering of therapeutic logic.
Innovation in Action: TPD’s Clinical Momentum and Strategic Breakthroughs
The field of Targeted Protein Degradation (TPD) has rapidly progressed from scientific curiosity to clinical cornerstone. What began as a visionary approach to hijack the cell’s proteostasis machinery is now reshaping therapeutic development across oncology, immunology, neurology, and beyond.
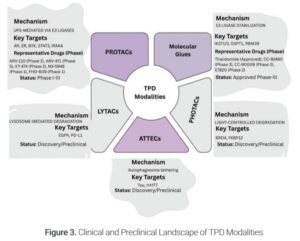
The momentum is undeniable. Researchers have now explored the degradation of over 130 protein targets, many of which were previously deemed “undruggable.” This surge in activity is translating into clinical progress, with more than 20 small-molecule degraders currently in clinical trials, a number that continues to grow as new modalities and indications emerge. These are not theoretical constructs—they represent a paradigm shift in drug development, replacing inhibition with targeted elimination of disease-driving proteins.
Kymera Therapeutics is extending Targeted Protein Degradation (TPD) beyond oncology, showcasing the technology’s versatility in tackling autoimmune and inflammatory disorders.
Their emphasis on degrading IRAK4 and STAT3 marks a significant advancement toward new treatments in these areas.

In parallel, Arvinas has provided crucial clinical validation for TPD with its PROTACs, particularly ARV-471, which continues to demonstrate promise in advanced-stage trials for ER+/HER2- breast cancer, underscoring the potential for effective and tolerable degrader-based therapies.
While ARV-110 has encountered challenges, the insights gained contribute to the overall progress of the field. Beyond these specific examples, TPD’s clinical footprint is rapidly expanding. Proteins previously regarded as intractable, involved in neurodegenerative diseases such as Tau, LRRK2, and TDP-43, are now viable targets. Hematologic malignancies are also witnessing advancements, with compounds like NX-5948 and BGB-16673 targeting BTK and BCL6 progressing through trials for lymphoma and leukemia. This widening clinical application reflects the diversification of TPD technologies, evolving beyond the foundational heterobifunctional PROTACs to encompass molecular glues for linker less degradation, LYTACs for targeting extracellular and membrane-bound proteins, and emerging platforms like ATTECs, AUTACs, and PHOTACs, which introduce innovative mechanisms such as autophagy and light activation. This multifaceted strategy highlights the adaptability and potential of TPD to revolutionize therapeutic interventions across a broad spectrum of diseases.
Coupled with these innovations is the integration of AI-driven drug discovery, which is accelerating the optimization of selectivity, potency, and pharmacokinetics. Tools like predictive modeling, degrader screening platforms, and AI-based linker design are pushing the boundaries of what is considered druggable.
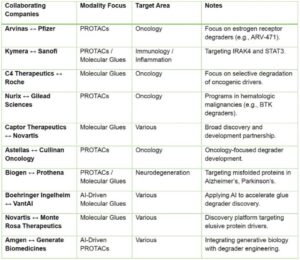
The strategic confidence in TPD is equally robust. Biotech pioneers such as Arvinas, Kymera, C4 Therapeutics, Nurix, and Vividion have inked multi-billion-dollar partnerships with industry leaders like Pfizer, Roche, Sanofi, and Genentech. These alliances reflect not only financial backing but a shared belief in TPD’s scalability, regulatory potential, and commercial viability.
Additional partnerships—such as those between Novartis and Captor Therapeutics, Bristol Myers Squibb and Synthego, and Astellas and Cullinan Oncology—further illustrate the modality’s strategic value, particularly in addressing challenging targets and complex disease biology.
The increasing number of clinical trials, bolstered by promising early-phase data, underscores TPD’s clinical relevance and translational strength. It is no longer a future-facing concept—it is a front-running modality in the next generation of precision therapeutics, redefining what’s possible in drug discovery and development.
Strategic Collaborations in TPDs: Key Industry Partnerships by Modality and Therapeutic Focus”
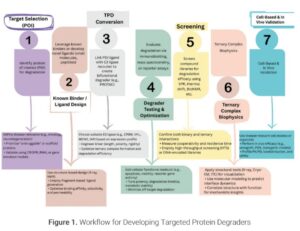
Challenges and the Path Forward for Targeted Protein Degraders
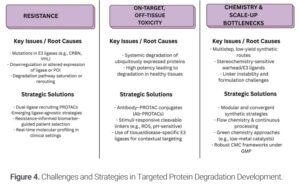
As targeted protein degradation (TPD) gains momentum across industry and academia, propelled by clinical-stage PROTACs and molecular glues, it’s easy to focus on the potential while underestimating the scientific and translational obstacles that still remain. Beneath the promise lies a web of unresolved issues—each one a critical frontier for innovation.
Despite the innovative nature of targeted protein degraders, the development of resistance is an emerging concern, observed in both laboratory and early clinical settings. Unlike traditional drugs where resistance stems from binding site mutations, degraders face a more complex landscape, with potential failure points at the E3 ligase, the targeted protein, or the cellular degradation machinery itself. Research indicates that resistance can arise from E3 ligase alterations, saturation of the degradation system by competing proteins, or changes in the target protein’s structure or location. To counter this, scientists are developing next-generation strategies, including dual-E3 ligase degraders and adaptable degrader libraries, alongside real-time biomarker monitoring for personalized treatment.
Another critical challenge is on-target, off-tissue toxicity. While degraders offer potent protein removal, this potency can lead to unintended side effects when the target protein is present in healthy tissues. Systemic degradation of essential proteins may result in dose-limiting toxicities. To mitigate this, researchers are exploring antibody-targeted degraders and stimuli-responsive linkers that confine activity to diseased tissues. The ultimate goal is context-specific degradation, tailoring the therapy to the disease environment.
Finally, the complex chemical synthesis of degraders presents a major bottleneck for large-scale production. These molecules often require lengthy, multi-step synthesis, precise stereochemical control, and optimized linkers. Variations in the synthesis can lead to significant differences in product quality, and the production of crucial ligands remains challenging. To overcome this, the field must prioritize modular chemistry, flow chemistry, and greener synthetic methods. Furthermore, evolving regulatory expectations for these complex molecules necessitate the development of robust and scalable manufacturing processes.
Are PROTACs the New “Magic Bullets” for Infectious Diseases?
The century-old concept of a “magic bullet,” first proposed by Paul Ehrlich to describe treatments that could selectively eliminate pathogens without harming healthy tissues, is being redefined through the emerging science of targeted protein degradation (TPD).
With the advent of PROTACs and molecular glues, modern medicine now possesses tools capable of degrading disease-driving proteins with remarkable precision by co-opting the cell’s own ubiquitin-proteasome system.
This approach represents more than a novel mechanism—it marks a fundamental shift in therapeutic logic. By catalytically eliminating rather than simply inhibiting proteins, degraders offer a powerful strategy to overcome drug resistance. This has already shown promise in treating drug-resistant viral infections such as hepatitis C and influenza. Moreover, TPD opens new avenues for tackling previously “undruggable” proteins, including transcription factors and scaffolding proteins, which have historically eluded conventional small molecule therapies. Intriguingly, the strategy also allows researchers to revisit and repurpose previously abandoned drug candidates by transforming weak inhibitors into potent degraders.
Yet, as with any transformative technology, challenges remain. Resistance mechanisms such as changes in E3 ligase expression or degradation pathway saturation are emerging, calling for innovative countermeasures like dual-ligase PROTACs and patient stratification based on biomarker profiling. Concerns about off-tissue toxicity are being addressed through antibody–PROTAC conjugates and linkers that respond to environmental cues such as pH or light, offering more refined spatial and temporal control. Meanwhile, advances in synthetic chemistry and AI-driven design are beginning to simplify the development of these complex molecules, reducing barriers to scalability and clinical translation.
The growing interest in TPD is mirrored by significant commercial activity. Strategic alliances—such as those between Arvinas and Pfizer or Kymera and Sanofi—and venture funding exceeding $700 million in recent years underscore the sector’s momentum. Companies like Nurix and C4 Therapeutics are making progress on degraders for oncology and neurodegenerative disorders, while emerging platforms like LYTACs and PHOTACs are extending the reach of degradation technologies to extracellular proteins and light-activated settings.
Looking ahead, TPD is evolving into a programmable platform for therapeutic intervention. New modalities such as RNA-targeting degraders (RIBOTACs) and tissue-specific E3 ligase recruitment strategies are pushing the boundaries of what’s possible. As degraders begin to enter combination regimens with immunotherapies or gene editing tools, their catalytic, precise, and durable mechanism of action promises to deliver synergistic benefits across a wide range of diseases.
In essence, targeted protein degradation has matured from an exciting concept to a tangible therapeutic revolution. What began as Ehrlich’s metaphorical bullet has transformed into a sophisticated molecular toolkit—one capable not only of targeting but of rewriting the biological narrative of disease.
We are no longer merely blocking proteins—we are reengineering the way diseases are treated at their source.

References and notes
- Arvinas. (2024). Clinical progress of ARV-110 and ARV-471. Retrieved from https://ir.arvinas.com/news-releases
- Churcher, I. (2018). PROTAC-induced protein degradation in drug discovery: Breaking the rules or just making new ones? Journal of Medicinal Chemistry, 61(2), 444–452. https://doi.org/10.1021/acs.jmedchem.7b01229
- Crews, C. M., Deshaies, R. J., & Sakamoto, K. M. (2001). Targeted protein degradation and the development of PROTACs. Journal of Biological Chemistry, 276(30), 28320–28323. https://doi.org/10.1074/jbc.M102295200
- GlobalData. (2023). Investment landscape in targeted protein degradation. Retrieved from https://njbio.com/targeted-protein-degraders
- Kymera Therapeutics. (2024). Pipeline overview. Retrieved from https://investors.kymeratx.com
- Nurix Therapeutics. (2024). NX-2127 and NX-5948: Phase 1 clinical trial data. Retrieved from https://www.nurixtx.com
- Novo Nordisk & Neomorph. (2024, February). Multi-target collaboration for molecular glue degraders. Retrieved from https://www.prnewswire.com/news-releases/neomorph-announces-multi-target-collaboration-with-novo-nordisk
- Novartis & Monte Rosa Therapeutics. (2024, October). Global license agreement for MRT-6160. Retrieved from https://ir.monterosatx.com
- Pfizer & Arvinas. (2021, July). Global collaboration to develop ARV-471. Retrieved from https://www.pfizer.com/news/press-release-detail/arvinas-and-pfizer-announce-global-collaboration-develop
- Zhou, P., Wang, W., Huang, S., & Wang, X. (2021). Linker design in PROTACs: Insights from structure-based optimization strategies. European Journal of Medicinal Chemistry, 216, 113308. https://doi.org/10.1016/j.ejmech.2021.113308
Kishore Hotha, PhD, MBA
President, Dr. Hotha’s Life Sciences LLC
Dr. Kishore Hotha is a distinguished leader in the pharmaceutical biotech and CDMO sectors, with a strong track record in advancing drug substance and product development across small and large molecules, including Antibody-Drug Conjugates (ADCs), oligonucleotides, peptides, and complex formulations. Throughout his career, he has been pivotal in the submission of numerous INDs, NDAs, and ANDAs, guiding these projects from concept to commercialization. Currently, Dr. Hotha is the President of Dr. Hotha’s Life Sciences LLC, a consulting firm dedicated to simplifying complex drug development challenges. Previously, he served as Global Vice President at Veranova, where he led analytical research and development operations, and held significant leadership roles at Lupin and Dr. Reddy’s Laboratories. With over 50 publications and several editorial board positions, Dr. Hotha remains an influential figure in shaping industry standards and advancing pharmaceutical innovation.


What complexities are associated with developing CMC (Chemistry, Manufacturing, and Controls) for Heterobifunctional Degraders (HBDs)?
Heterobifunctional Degraders (HBDs), which offer a transformational approach to positively impacting human health, pose exciting opportunities and challenges for both Drug Substance (DS) and Product (DP) development and manufacturing. Herein we will consider DS.
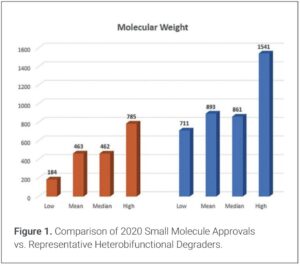
A fundamental consideration for DS development, manufacturing, and controls for HBDs is that while they are small molecules and once developed have inherent manufacturing and supply chain benefits compared to other common modalities (1), they are larger than most small molecule pharmaceuticals. Figure 1 depicts a molecular weight (MW) comparison of forty small molecules approved in 2020 vs. forty representative HBDs (2). While the mean and median MWs of the degraders are unsurprisingly about twice that of standard small molecules, it is notable that standard small molecules can approach the average MW of HBDs. Nonetheless, HBDs inherently have high MWs that can be triple the size of a standard small molecule.
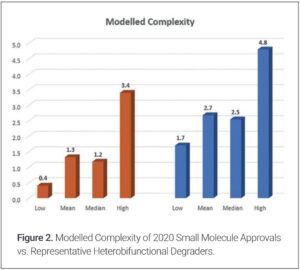
As the overall complexity of a DS is comprised of more than the MW, these compounds were assessed using a simple complexity model incorporating MW, number of rings, and number of controlled stereocenters (3). Figure 2 indicates a similar trend to the MW assessment.
Additionally, potentially less obvious considerations include understanding and control of physical attributes, and analytical method development, specifically chromatography.
In general, the most practical and scalable method for isolating and purifying small molecules is crystallization. For a crystallization, one must first have identified at least one crystalline form, either as free base/acid, salt, or cocrystal (4).
There is no guarantee, however, that a given compound will have a stable crystalline form. It is possible to develop an amorphous precipitation, however this is often non-trivial, as it will typically have less impurity purging power than a crystallization, and may have unfavorable physical attributes (5).
Alternatively, larger small molecules often have intricate polymorphic landscapes and crystallization processes (6), which can complexify CMC development.
The development and validation of a robust analytical method that is capable of separating impurities from the DS and other impurities is critical to ensure purity and potency. The MW of HBDs can result in challenging chromatography development, as impurities often have very similar chemical properties as the DS (e.g., a diastereomer).
With respect to chemical stability, it is good practice to assess potential solid phase and solution phase degradation pathways at the discovery stage. Many published HBDs contain a glutarimide ring which is hydrolytically labile and if developed as a single isomer, epimerizable. The fact that HBDs are large results in a higher chance of a functional group sensitive to common causes of degradation. Consequently, it is advisable to have an early stability-indicating chromatography method available for the stress studies.
Owing to the larger MW of HBDs, in some cases standard scale up challenges may be exacerbated and on average will require more complex route scouting and involve a larger number of synthesis steps. This will often result in more development required prior to scale up, complex supply chains, higher costs, and longer lead times.
Finally, as with all New Chemical Entities, one should have a toxicological industrial hygiene assessment performed to ensure safe handling in the laboratory and manufacturing facility (7).
What factors should a Biotech company consider when selecting a CDMO for Heterobifunctional Degrader (HBD) development, particularly concerning expertise in bifunctional molecule handling?
As discussed above, HBDs are small molecules, albeit typically larger and more complex. In some cases, HBDs may be highly potent and require appropriate handling procedures and controls to ensure operator safety; if this is the case, partners with capabilities and expertise in handling substances at the determined OEL/OEB should be selected. Consequently, it is recommended that an OEL/OEB assessment is made as early as feasible. Outside of this, the selection of partners for development and/or manufacturing are not fundamentally different from standard small molecules.
That stated, HBDs typically require a higher level of process development, analytical development, and technology realization in a pilot plant. It is important for Biotechs interested in rapidly bringing their innovative therapies to patients to engage with CROs and CDMOs with appropriate scientific expertise, technologies, communication skills, manufacturing capabilities, quality assurance practices, and culture. As the availability of DS is often rate limiting for IND-enabling toxicology and early clinical studies, the below considerations are mainly directed towards early deliveries.
As development necessarily precedes manufacturing, the first question a biotech needs to address is where the development will be performed – should one partner with a single, fully integrated CDMO to manage the entire delivery from route scouting through development to manufacturing, or should one engage multiple CROs and CDMOs? Partnering with a single company allows for efficiencies such as minimal technology transfers and enabling development of a process that aligns with the CDMO’s manufacturing capabilities. Utilizing multiple CROs/CDMOs could allow one to optimize the route, process, and controls by leveraging different technologies, provide geographic and financial flexibility, and manage risk to allow for business continuity, should issues arise.
As HBDs typically require more development and manufacturing, it is advisable to utilize partners who can apply state of the art expertise and equipment to accelerate the final delivery from initial development. While this approach will likely incur higher development costs up front, these are typically offset by a lower manufacturing cost and shorter delivery timelines. This includes access to and proficiency with High Throughput Experimentation (HTE), biocatalysis, continuous processing, crystallization development, analytical development, and knowledge management. The following paragraphs address some considerations for these capabilities.
HTE is a powerful tool for multiple reaction classes, particularly transition metal mediated transformations, but also for other reactions and processes, such as peptide couplings, chiral salt resolutions, biocatalysis, and resin screening for metal reduction (8). As many HBDs will utilize one or more of the above common reactions/ operations, access to HTE expertise and equipment can drastically speed development and potentially lower costs (e.g., by lowering a palladium (Pd) load or replacement of Pd with a non-precious metal) (9).
Continuous processing (CP) technologies, such as plug flow reactors (PFR) and continuous stirred tank reactors (CSTRs) have opened many reaction classes that were previously challenging to scale (10). Examples include hazardous chemistry, cryogenic reactions, photochemistry, and less obviously, kinetically fast reactions that rapidly decompose if not quickly quenched. In years past, most of these reactions would have required either significant development, or possibly a complete change in route, thereby lengthening the lead time to the final delivered DS. Note that while CP is becoming more common, not all CROs and CDMOs have adequate development and manufacturing expertise and equipment.
Beyond standard initial salt and form screening to identify candidate forms for development, it can be advisable to engage CROs/CDMOs who have expertise in developing data-driven isolation processes (11). This is often an underappreciated and misunderstood aspect of early development but is critical to ensure the final process can deliver the final DS and intermediates with acceptable quality and physicochemical characteristics.
Also discussed previously is the importance of sophisticated analytical chromatography development. High peak capacity methods will often be required to separate structurally related impurities from the DS; as HBDs have inherently high MWs, many of these impurities will chromatographically behave very similarly to the DS.
A final aspect a Biotech should consider is knowledge management. This includes both GMP documentation (e.g., executed batch records) and process development. Good documentation is critical to ensure compliance, to support regulatory filings, capture intellectual property, and to identify key areas for additional development as a product moves towards commercialization. It is a lighter lift to document and organize knowledge as it is generated than to later try recalling where a key document is or why a certain decision was made (e.g. route selection, catalytic reaction conditions).
In conclusion, while approaches to the development and manufacturing of HBDs are fully tractable and not fundamentally different from that of typical small molecules, their increased complexity brings a higher development and manufacturing burden to bear. This is particularly acute in early development when the availability of DS is often rate-liming to critical nonclinical and clinical development; long term manufacturing can be managed by proactive planning. The early selection of appropriate CRO/ CDMO partner(s) and establishing an effective collaboration will have a significant impact on the quality, speed, cost, and success of an HBD development program.
References and notes
- Clifford, K.A.; Levine, A.A.; Enright, D.E.; Neumann, P.J.; Chambers, J.D. Health Affairs, 2024, 43, 1546-1552.
- The 40 small molecules approved in 2020 were taken from Flick, Andrew C.; Leverett, Carolyn A.; Ding, Hong X.; McInturff, Emma L.; Fink, Sarah J.; Mahapatra, Subham; Carney, Daniel W.; Lindsey, Erick A.; DeForest, Jacob C.; France, Scott P.; Berritt, Simon; Bigi-Botterill, Simone V.; Gibson, Tony S.; Watson, Rebecca B.; Liu, Yiyang; O’Donnell, Christopher J. J. Med. Chem. 2022, 65, 14, 9607-9661. Antibody drug conjugate drug linkers were excluded. The 40 representative heterobifunctional degraders include published compounds from Arvinas, Dialectic Therapeutics, Nurix Therapeutics, Bristol Myers Squibb, C4 Therapeutics, Foghorn Therapeutics, and published and unpublished internal compounds from Kymera Therapeutics.
- The model assumes an average MW of 525 (50% weighting), average number of rings of 3.6 (25% weighting), and average number of stereocenters as 1.1 (25% weighting). Examples of more intricate complexity models include a) Krzyzanowski, A.; Pahl, A.; Grigalunas, M.; Waldmann, H. J. Med. Chem., 2023, 66, 18, 12739-12750. b) Karageorgis, G.; Douglas, J.J.; Howell, G.P. Org. Process Res. Dev. 2024, 28,8, 3339-3346. c) Jun, L.; Eastgate, M.D. Org. Biomol. Chem. 2015, 13, 26, 7164-7176. d) Coley, C. W.; Rogers, L.; Green, W.H.; Jensen, K. F. J. Chem. Inf. Model. 2018, 58, 2, 252-261. e) Wright, B.S.; Sarpong, R. Nat. Chem. Rev. 2024, 8, 776-792.
- Saha, A.; Ahangar, A.A.; Dar, A.A.; Thirunahari, S.; Parambil, J.V. Cryst. Growth Des. 2003, 23, 7558-7581.
- Zarkadas, D.; Pridgen, C.S.; Liotta, V. Journal of Pharmaceutical Sciences 2018, 107, 217-230.
- Yu, L.; Reuzel-Edens, S.M.; Mitchell, C.A. Org. Process Res. Dev. 2000, 4, 396-402.
- a) Ader, A.W.; Farris, J.P; Ku, R.H. Chemical Health and Safety 2005, 12, 4, 20-26.
- a) Mennen, S.M.; Alhambra, C.; Allen, C.L.; Barberis, M.; Berritt, S.; Brandt, T.A.; Campbell, A.D.; Castanon, J.; Cherney, A.H.; Christensen, M.; Damon, D.B.; de Diego, J.E.; Garcia-Cerrada, S.; Garcia-Losada, P.; Haro, R.; Janey, J.; Leitch, D.C.; Li, L.; Liu, F.; Lobben P.C.; MacMillan, D.W.C.; Magano, J.; McInturff, E.; Monfette, S.; Post, R.J.; Schultz, D.; Sitter, B.J.; Steven, J.M.; Strambeanu, J.I.; Twilton, J.; Wang, K.; Zajac, M.A. Org. Process Res. Dev. 2019, 23, 1213-1242. b) Welch, C.J.; Albaneze-Walker, J.; Leonard, W.R.; Biba, M.; DaSilva, J.; Henderson, D.; Laing, B.; Mathre, D.J.; Spencer, S.; Bu, X.; Wang, T. Org. Process Res. Dev. 2005, 9, 198-205. e) France, S.P.; Lewis, R.D.; Martinez, C.A. JACS AU, 2023, 3, 715-735.
- a) Wu, H.; Qu, B.; Nguyen, T.; Lorenz, J.C.; Buono, F.; Hadda, N. Org. Process Res. Dev. 2022, 26, 2281-2310. b) Chirik, P.J.; Engle, K.M.; Simmons, E.M.; Wisniewski, S.R. Org. Process Res. Dev. 2023, 27, 1160-1184.
- Cole, K.P.; Jaworski, J.N.; Kappe, C.O.; Kobayashi, S.; Maguire, A.R.; O’Kearney-McMullan, A.; Pesti, J. Org. Process Res. Dev. 2024, 28, 1269-1271.
- Paul, E.L.; Tung, H.-H.; Midler, M. Powder Technology, 2005, 150, 2, 133-143.
–

CMC Requirements: What complexities are associated with developing CMC (Chemistry, Manufacturing, and Controls) strategies for PROTACs, particularly around stability and scalability?
Developing CMC (Chemistry, Manufacturing, and Controls) strategies for PROTACs present several complexities, particularly around stability and scalability. These challenges arise due to the typically increased complicated molecular structure which would likely require a more complicated synthesis and the characteristics of PROTACs which tend to be less crystalline due to molecular size.
PROTACs are often composed of multiple functional groups connected by linkers, which possess functional groups that can be susceptible to chemical degradation. Ensuring early investment in forced degradation studies to understand the chemical stability and their degradation congeners of these molecules during synthesis, and storage, could stave off longer term development challenges. Accompanying these studies, comprehensive analytical methods are needed to characterize PROTACs, including their structure, and purity which can be time-consuming.
The synthesis of PROTACs often involves multiple steps, including the conjugation of different chemical moieties. With any lengthier synthesis, there is a higher level of focus on the optimization of yield, and ensuring reproducibility as cost, both with respect to monetary and time, will come heavily into play on the drug substance side, even at early stages of the program.
While not an issue in every case, given the molecular size of typical lead PROTACs, a hard look into its polymorphic landscape is particularly important as PROTACs often have poor solubility and bioavailability due to their size and hydrophilicity. Formulation strategies, such as the use of solubilizing agents, employing the use of a spray dried dispersion, or even prodrug approaches, may be necessary to improve these properties. Like the challenges encountered on the drug substance side, ensuring the stability of PROTACs in their final formulation is critical over its projected shelf life.
Scale-Up Challenges: What unique challenges do CDMOs face in scaling up PROTAC production from bench to commercial scale, and how can companies address these issues early on?
Scaling up a PROTAC from bench to commercial scale in principle has many similarities to the challenges faced when a highly complicated small molecule is nominated for development. In both these cases, the complexity of the synthesis is the central core considered. A focus into analytical chemistry will be crucial, where the ability to monitor the behavior of key transformations, by ensuring the robust separation of impurities on an analytical method are critical in guiding process chemistry development.
–

The Evolution of Synthetic Drugs: Navigating the Complexity of Targeted Protein Degraders
We are at the dawn of a new era of synthetic drug development where novel chemical modalities, including targeted protein degraders (TPD), are emerging as an exciting class of potential therapeutics. The field of targeted protein degradation began somewhat serendipitously with the discovery of molecular glues, small molecules that induce protein-protein interactions between a target protein and an E3 ubiquitin ligase. One of the notable early examples is thalidomide, which was later found to bind to cereblon (CRBN), a component of an E3 ubiquitin ligase complex, leading to the degradation of specific transcription factors (1). This unexpected mechanism explained the drug’s efficacy in treating multiple myeloma and led to the development of other immunomodulatory imide drugs (IMiDs) such as lenalidomide and pomalidomide. These molecular glues do not require a distinct linking moiety, offering favorable drug-like properties due to their smaller size compared to later-developed bifunctional degraders. Building on these insights, rationally designed Proteolysis Targeting Chimeras (PROTACs®) emerged in the early 2000s (2). These bifunctional molecules were engineered to harness the cell’s natural protein disposal system, the ubiquitin-proteasome pathway, to selectively eliminate disease-causing proteins.
Heterobifunctional protein degraders like PROTACs consist of three key components: a ligand that binds to the target protein, a linker, and a ligand that recruits an E3 ubiquitin ligase. As the PROTAC molecule binds to the target protein and the E3 ligase, a ternary complex is formed which leads to the ubiquitination and subsequent degradation of the target protein. After this protein is degraded, the PROTAC molecule is released and can repeat this process with another target protein. Unlike small molecule inhibitors that require a protein to possess a suitable binding pocket, targeted protein degraders may bind anywhere on the protein that is to be eliminated. This innovative approach opens new possibilities for addressing previously “undruggable” targets, making TPDs suitable for evaluation across a wide swath of the proteome and offering potential advantages over traditional inhibitors, such as the ability to target non-enzymatic proteins and a catalytic mode of action (3). This catalytic behavior means that TPDs can potentially be effective at lower concentrations compared to traditional inhibitors, which need to maintain a high occupancy of the target protein to be effective (4). Despite being conceptualized in 2001, it took nearly two decades for the first targeted protein degraders to enter clinical trials. In 2019, the first oral heterobifunctional protein degrader compounds began clinical evaluation in patients with advanced or metastatic cancer. A significant milestone was achieved in 2022 when an estrogen receptor degrader advanced to Phase III trials, marking a crucial validation of the targeted protein degrader approach (5).
This progress has catalyzed industry-wide interest, with over 20 TPD molecules entering clinical trials.
Challenges in Developing Targeted Protein Degraders
While targeted protein degraders hold immense promise, their development presents several significant challenges, including 1) low solubility and bioavailability, 2) complex and lengthy chemical syntheses, 3) formulation challenges (to enable oral administration) as well as 4) unique pharmacokinetic and pharmacodynamic considerations.
1. Low Solubility and Bioavailability
One of the primary hurdles in developing targeted protein degraders is their often-poor solubility and bioavailability. These molecules tend to be larger and more lipophilic than traditional small molecule drugs, which can potentially lead to reduced absorption in the gastrointestinal tract, limited distribution to target tissues, and increased likelihood of first-pass metabolism. Heterobifunctional protein degraders typically have molecular weights exceeding 800 Da and high topological polar surface areas, with most clinical-stage degraders violating at least four parameters of Lipinski’s Rule of 5 (6). They are generally categorized as BCS class IV compounds (low solubility and low permeability) or BCS class II compounds (low solubility and high permeability), presenting significant challenges for pharmaceutical development. Notwithstanding these obstacles, many clinical-stage TPDs are being developed for oral administration, highlighting the industry’s interest in this route despite the formulation challenges (7). To address these issues, scientists must carefully balance the physicochemical properties of the degrader molecules, often employing strategies such as reducing molecular weight, optimizing lipophilicity, incorporating solubilizing groups, or utilizing enabling formulations.
2. Complex and Lengthy Chemical Syntheses
The structural complexity of targeted protein degraders often necessitates long and difficult chemical syntheses. Accordingly, significant chemical process development is required to mitigate the potential challenges of low overall yields, increased production costs, and difficulties in scaling up for clinical trials and commercial manufacturing.
This synthetic complexity arises from:
1. The need for multiple functionalities within a single molecule
2. Stereospecific synthesis requirements
3. Challenging coupling reactions between the protein-binding ligand, linker, and E3 ligase recruiter
A constraint in TPD development so far has been the restricted range of available E3 ligase ligands. Despite the human genome containing over 600 E3 ligases, only a select few – primarily cereblon (CRBN) and von Hippel-Lindau (VHL) – have been successfully leveraged in TPD development (8). Moreover, the strategic design of linkers proves critical not merely for synthetic accessibility but also for facilitating effective ternary complex formation between the target protein and E3 ligase.
Linker Composition and Optimization
The linker component of TPDs, while often overlooked, is critical for both physicochemical properties and biological activity. Most successful TPDs utilize polyethylene glycol (PEG) chains, alkyl chains, or combinations of both (9). PEG linkers typically enhance aqueous solubility but may reduce cell permeability, while alkyl linkers can improve membrane permeation at the cost of solubility.
Studies have demonstrated that optimal linker length is highly target-dependent, typically ranging from 5-15 atoms.
Too short a linker prevents simultaneous binding of both proteins, while excessive length introduces entropic penalties that reduce potency (10).
The introduction of conformational constraints through rigid elements (e.g., alkynes, aromatics, cycloalkanes) can pre-organize the TPD in a productive conformation, enhancing potency and selectivity (11). Structural studies of TPD-mediated ternary complexes have revealed that linkers can influence not only the proximity of the target protein to the E3 ligase but also the specific protein-protein interactions that form between them, a phenomenon termed “cooperativity” (12). This understanding has led to more rational approaches to linker design, moving beyond simple trial-and-error to structure-guided optimization.
3. Formulation Challenges
The unique properties of targeted protein degraders present significant formulation challenges. Their poor aqueous solubility often necessitates the development of advanced formulation strategies to achieve adequate bioavailability and stability. Some approaches include:
Lipid-Based Formulations (LBFs): These formulations can improve solubility and enhance lymphatic uptake, potentially bypassing first-pass metabolism. Recent studies with model TPD compounds like BRD4 degraders have demonstrated >50-fold solubility enhancement in biorelevant media using self-emulsifying formulations (13). LBFs present the drug in solution pre- and/or post-administration, increasing oral absorption via solubility enhancement and sustaining supersaturation. It has also been demonstrated that LBFs can significantly improve the oral bioavailability of very poorly water-soluble drugs through enhanced solubilization and lymphatic transport (14).
Amorphous Solid Dispersions (ASDs): This technique involves dispersing the drug in a polymer matrix to stabilize the higher-energy amorphous form, providing both physical stability and enhanced dissolution. The most common industrial methods for scaling ASDs are spray drying and hot melt extrusion. For several TPD compounds, ASDs with different polymers at varying drug loadings have been evaluated with variable success (15). One advantage of some TPD compounds is their tendency to be naturally amorphous due to their larger size and structural flexibility, potentially simplifying ASD development. For early development work when TPDs may only be available in limited quantities, novel screening tools for ASD formulations have been developed including vacuum compression molding approaches (16).
Novel Combination Approaches: Emerging research explores combining LBFs and ASDs to maximize solubility and supersaturation enhancement, such as adding surfactants to ASDs (15c) or polymers to LBFs, potentially addressing the dual challenges of solubility and permeability. For example, polymeric precipitation inhibitors have been shown to promote supersaturation and enhance drug absorption from lipid-based formulations (17).
Drug Nanoparticles: Amorphous drug nanoparticles offer another potential approach for TPDs or other poorly soluble compounds, with nanomilling or nanoprecipitation methods providing materials with improved dissolution profiles. For example, a scalable approach for formulating fast-dissolving nanoparticles that significantly enhanced dissolution rates and apparent solubility was recently reported using Flash NanoPrecipitation (FNP). The integrated process involves nanoparticle formation, concentration by tangential flow ultrafiltration, and then spray drying to obtain a powder (18).
Cyclodextrin Complexation: Forming inclusion complexes with cyclodextrins can potentially enhance the solubility and stability of degrader molecules. Among the various pharmaceutical applications of cyclodextrins, their ability to form inclusion complexes with hydrophobic drugs has been shown to be an attractive option for increasing their apparent water solubility (19).
Each of these formulation strategies comes with its own set of opportunities and challenges, offering pathways to address stability issues, potential for drug-excipient interactions, and scalability.
4. Pharmacokinetic and Pharmacodynamic Considerations
The complex nature of targeted protein degraders introduces unique pharmacokinetic and pharmacodynamic challenges:
Tissue Distribution: Ensuring sufficient concentrations of the degrader reach the intended target tissues can be challenging, particularly for targets in the central nervous system or other poorly perfused areas.
Protein Degradation Kinetics: The catalytic nature of protein degradation introduces complexities in dose-response relationships and requires careful consideration of degradation rates, protein resynthesis, and potential feedback mechanisms (20).
Drug-Drug Interactions: The reliance on the cellular protein degradation machinery may raise concerns about potential drug-drug interactions, particularly with other drugs that modulate the ubiquitin-proteasome system.
“Molecular Chameleon” Behavior: TPDs can flexibly adapt to the intestinal environment for epithelial permeation, which may contribute to their sometimes better-than-expected oral bioavailability despite poor physicochemical properties (21).
Biorelevant Solubility Testing: The importance of testing in fasted-state simulated intestinal fluid (FaSSIF) and fed-state simulated intestinal fluid (FeSSIF) is a key parameter for optimizing oral absorption of TPD compounds, since permeability is difficult to improve for inherently large and flexible molecules (22), and biorelevant solubility may be significantly higher in FeSSIF than in aqueous buffered (pH 7) solutions (7).
Emerging Chemical TPD Modalities
The field of targeted protein degradation continues to evolve rapidly, with innovative approaches expanding beyond heterobifunctional degraders. Accordingly, the protein degrader category has grown to include not only molecular glues and TPDs but also:
RIPTACs (Regulated Induced Proximity Targeting Chimeras) where one end of the heterobifunctional molecule binds a ‘target protein’ (e.g. in cancer cells) and the other binds an ‘essential protein’ that is required for cell survival.
Accordingly, when cells carry both proteins, the RIPTAC brings them together in a complex which silences the essential protein and kills the cancer cells (23).
Covalent PROTACs incorporate reactive groups that form irreversible bonds with their target proteins, potentially enabling more efficient degradation of proteins with transient binding sites or low expression levels. This approach may overcome the challenge of achieving sufficient target occupancy with reversible binders (24).
Photoswitchable degraders like PHOTACs that incorporate light-sensitive moieties which allow for spatial and temporal control of degradation activity. These tools are particularly valuable for studying protein function in specific cellular compartments or developmental stages (25).
Heterobifunctional degraders beyond the ubiquitin-proteasome system continue to expand, including AUTACs that direct proteins to the autophagy pathway, LYTACs that utilize the lysosomal degradation pathway, and MADTACs (Macroautophagy Degradation Targeting Chimeras) that harness selective autophagy mechanisms (26). These approaches offer alternative solutions for proteins that are poor substrates for proteasomal degradation.
Conclusion
The development of targeted protein degraders represents a significant advance in the pursuit of more precise and effective therapies. However, the increased molecular complexity of these agents presents formidable challenges in their design, synthesis, formulation, and clinical development. Overcoming these hurdles requires interdisciplinary collaboration among chemists, chemical engineers, formulation scientists, and clinical researchers. As the field continues to evolve, innovative approaches to synthesis, formulation, and delivery will be crucial in realizing the full potential of targeted protein degraders. The progress from concept to clinic, with a growing wave of targeted protein degraders now entering clinical trials, clearly demonstrates the momentum in the field. Beyond oncology, there is significant potential for expansion into neurodegenerative diseases, metabolic disorders, viral infections, and inflammatory conditions with the hope of potentially revolutionizing the treatment of a wide range of diseases (27).
References and notes
- Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345-1350.
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001;98(15):8554-8559.
- ~85% of the proteome is considered ‘undruggable’ by traditional small-molecule inhibitors, see: Neklesa TK, Winkler JD & Crews CM Targeted protein degradation by PROTACs. Pharmacology & Therapeutics 2017;174 :138–144. Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, Zinn N, Grandi P, Shimamura S, Bergamini G, Faelth-Savitski M, Bantscheff M, Cox C, Gordon DA, Willard RR, Flanagan JJ, Casillas LN, Votta BJ, den Besten W, Famm K, Kruidenier L, Carter PS, Harling JD, Churcher I, Crews CM. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11(8):611-617.
- Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21(3):181-200.
- Chirnomas D, Hornberger KR, Crews CM. Protein degraders enter the clinic – a new approach to cancer therapy. Nat Rev Clin Oncol. 2023;20(4):265-278.
- Edmondson SD, Yang B, Fallan C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: recent progress and future challenges. Bioorg Med Chem Lett. 2019;29(15):1555-1564. Lipinski’s rule-of-five serves as a conceptual framework to assess if a compound would be suitable for oral delivery and in-cludes (i) molecular weight less than 500 g/mol, (ii) no more than five H-bond donors, (iii) no more than ten H-bond acceptors and (iv) an octanol/water partition coeficient (logP) of < 5.
- Yang W, Saboo S, Zhou L, Askin S, Bak A. Early evaluation of op-portunities in oral delivery of PROTACs to overcome their molecular challenges. Drug Discov Today. 2024;29(2):103865.
- Belcher BP, Ward CC, Nomura DK. Ligand ability of E3 ligas-es for targeted protein degradation applications. Biochemistry. 2023;62(4):588-600.
- Troup RI, Fallan C, Baud MGJ. Current strategies for the design of PROTAC linkers: a critical review. Explor Target Antitumor Ther. 2020;1(5):273-312.
- Zorba A, Nguyen C, Xu Y, Starr J, Borzilleri K, Smith J, Zhu H, Farley KA, Ding W, Schiemer J, Feng X, Chang JS, Uccello DP, Young JA, Garcia-Irrizary CN, Czabaniuk L, Schuff B, Oliver R, Montgomery J, Hayward MM, Coe J, Chen J, Niosi M, Luthra S, Shah JC, El-Kattan A, Qiu X, West GM, Noe MC, Shanmugasundaram V, Gilbert AM, Brown MF, Calabrese MF. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci USA. 2018;115(31):E7285-E7292.
- Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safa-ee N, Jedrychowski MP, Ponthier CM, Ishoey M, Zhang T, Mancias JD, Gray NS, Bradner JE, Fischer ES. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol. 2018;14(7):706-714.
- Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M, Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13(5):514-521.
- Rathod D, Fu Y, Patel K. BRD4 PROTAC as a novel therapeutic ap-proach for the treatment of vemurafenib resistant melanoma: Pre-formulation studies, formulation development and in vitro evalua-tion. Eur J Pharm Sci. 2019;138:105039.
- Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based for-mulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231-248.
- (a) Pöstges F, Kayser K, Appelhaus J, Monschke M, Gütschow M, Steinebach C, Wagner KG. Solubility enhanced formulation ap-proaches to overcome oral delivery obstacles of PROTACs. Phar-maceutics. 2023; 15(1):156. (b) Hofmann N, Harms M, Mader K. ASDs of PROTACs: Spray-dried solid dispersions as enabling formu-lations. International Journal of Pharmaceutics. 2024;650:123725. (c) Zhang H, Wu H, Wang L, Gao Y, Machín Galarza L, Zhao Y, Wang Z, Gao L, Han J. Preparation and characterization of ASDs improves the solubility and dissolution performance of a PROTAC drug. Jour-nal of Drug Delivery Science and Technology 2025;107:106837.
- Shadambikar G, Kipping T, Di-Gallo N, Elia A-G, Knüttel A-N, Treffer D, Repka MA. Vacuum compression molding as a screening tool to investigate carrier suitability for hot-melt extrusion formulations. Pharmaceutics. 2020;12(11):1019.
- Suys EJA, Chalmers DK, Pouton CW, Porter CJH. Polymeric precip-itation inhibitors promote fenofibrate supersaturation and enhance drug absorption from a type IV lipid-based formulation. Mol Pharm. 2018;15(6):2355-2371.
- Armstrong M, Wang L, Ristoph K, Tian C, Yang J, Ma L, Panmai S, Zhang D, Nagapudi K, Prud’homme RK. Formulation and scale-up of fast-dissolving lumefantrine nanoparticles for oral malaria therapy. J Pharm Sci. 2023;112(8):2267-2275.
- Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins: basic science and product development. J Pharm Pharmacol. 2010;62(11):1607-1621.
- Churcher I. Protac-Induced Protein Degradation in Drug Discov-ery: Breaking the Rules or Just Making New Ones? J Med Chem. 2018;61(2):444-452.
- Poongavanam V, Kihlberg J. PROTAC cell permeability and oral bioavailability: a journey into uncharted territory. Future Med Chem. 2022;14(2):123-126.
- Cantrill C, Chaturvedi P, Rynn C, Petrig Schaffland J, Walter I, Wittwer MB. Fundamental aspects of DMPK optimization of targeted protein degraders. Drug Discov Today. 2020;25(6):969-982.
- Raina K, Forbes CD, Stronk R, Rappi Jr. JP, Eastman KJ, Zaware N, Yu X, Li H, Bhardwaj A, Gerritz SW, Forgione M, Hundt A, King MP, Posner ZM, Correia AD, McGovern A, Puleo DE, Chenard R, Mous-seau JJ, Vergara JI, Garvin E, Macaluso J, Martin M, Bassoli K, Jones K, Garcia M, Howard K, Yaggi M, Smith LM, Chen JM, Mayfield AB, De Leon CA, Hines J, Kayser-Bricker KJ, Crews CM. Regulated induced proximity targeting chimeras-RIPTACs-A heterobifunctional small molecule strategy for cancer selective therapies. Cell Chemical Biology. 2024;31:1490–1502.
- Gabizon R, Shraga A, Gehrtz P, Livnah E, Shorer Y, Gurwicz N, Avram L, Unger T, Aharoni H, Albeck S, Brandis A, Shulman Z, Katz B-Z, Herishanu Y, London N. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J Am Chem Soc. 2020;142(27):11734-11742.
- Reynders M, Matsuura BS, Bérouti M, Simoneschi D, Marzio A, Pagano M, Trauner D. PHOTACs enable optical control of protein degradation. Sci Adv. 2020 Feb21;6(8):eaay5064.
- Alabi SB, Crews CM. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021;296:100647.
- Zhong G, Chang X, Xie W, Zhou X. Targeted protein degradation: advances in drug discovery and clinical practice. Sig Transduct Target Ther. 2024;9:308.

